{"title":"通过晶体学、核磁共振和MD模拟揭示了Eph受体-配体界面的蛋白质动力学。","authors":"Haina Qin, Liangzhong Lim, Jianxing Song","doi":"10.1186/2046-1682-5-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The role of dynamics in protein functions including signal transduction is just starting to be deciphered. Eph receptors with 16 members divided into A- and B- subclasses are respectively activated by 9 A- and B-ephrin ligands. EphA4 is the only receptor capable of binding to all 9 ephrins and small molecules with overlapped interfaces.</p><p><strong>Results: </strong>We first determined the structures of the EphA4 ligand binding domain (LBD) in two crystals of P1 space group. Noticeably, 8 EphA4 molecules were found in one asymmetric unit and consequently from two crystals we obtained 16 structures, which show significant conformational variations over the functionally critical A-C, D-E, G-H and J-K loops. The 16 new structures, together with previous 9 ones, can be categorized into two groups: closed and open forms which resemble the uncomplexed and complexed structures of the EphA4 LBD respectively. To assess whether the conformational diversity over the loops primarily results from the intrinsic dynamics, we initiated 30-ns molecular dynamics (MD) simulations for both closed and open forms. The results indicate that the loops do have much higher intrinsic dynamics, which is further unravelled by NMR H/D exchange experiments. During simulations, the open form has the RMS deviations slightly larger than those of the closed one, suggesting the open form may be less stable in the absence of external contacts. Furthermore, no obvious exchange between two forms is observed within 30 ns, implying that they are dynamically separated.</p><p><strong>Conclusions: </strong>Our study provides the first experimental and computational result revealing that the intrinsic dynamics are most likely underlying the conformational diversity observed for the EphA4 LBD loops mediating the binding affinity and specificity. Interestingly, the open conformation of the EphA4 LBD is slightly unstable in the absence of it natural ligand ephrins, implying that the conformational transition from the closed to open has to be driven by the high-affinity interaction with ephrins because the weak interaction with small molecule was found to be insufficient to trigger the transition. Our results therefore highlight the key role of protein dynamics in Eph-ephrin signalling and would benefit future design of agonists/antagonists targeting Eph receptors.</p>","PeriodicalId":9045,"journal":{"name":"BMC Biophysics","volume":"5 ","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2046-1682-5-2","citationCount":"23","resultStr":"{\"title\":\"Protein dynamics at Eph receptor-ligand interfaces as revealed by crystallography, NMR and MD simulations.\",\"authors\":\"Haina Qin, Liangzhong Lim, Jianxing Song\",\"doi\":\"10.1186/2046-1682-5-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The role of dynamics in protein functions including signal transduction is just starting to be deciphered. Eph receptors with 16 members divided into A- and B- subclasses are respectively activated by 9 A- and B-ephrin ligands. EphA4 is the only receptor capable of binding to all 9 ephrins and small molecules with overlapped interfaces.</p><p><strong>Results: </strong>We first determined the structures of the EphA4 ligand binding domain (LBD) in two crystals of P1 space group. Noticeably, 8 EphA4 molecules were found in one asymmetric unit and consequently from two crystals we obtained 16 structures, which show significant conformational variations over the functionally critical A-C, D-E, G-H and J-K loops. The 16 new structures, together with previous 9 ones, can be categorized into two groups: closed and open forms which resemble the uncomplexed and complexed structures of the EphA4 LBD respectively. To assess whether the conformational diversity over the loops primarily results from the intrinsic dynamics, we initiated 30-ns molecular dynamics (MD) simulations for both closed and open forms. The results indicate that the loops do have much higher intrinsic dynamics, which is further unravelled by NMR H/D exchange experiments. During simulations, the open form has the RMS deviations slightly larger than those of the closed one, suggesting the open form may be less stable in the absence of external contacts. Furthermore, no obvious exchange between two forms is observed within 30 ns, implying that they are dynamically separated.</p><p><strong>Conclusions: </strong>Our study provides the first experimental and computational result revealing that the intrinsic dynamics are most likely underlying the conformational diversity observed for the EphA4 LBD loops mediating the binding affinity and specificity. Interestingly, the open conformation of the EphA4 LBD is slightly unstable in the absence of it natural ligand ephrins, implying that the conformational transition from the closed to open has to be driven by the high-affinity interaction with ephrins because the weak interaction with small molecule was found to be insufficient to trigger the transition. Our results therefore highlight the key role of protein dynamics in Eph-ephrin signalling and would benefit future design of agonists/antagonists targeting Eph receptors.</p>\",\"PeriodicalId\":9045,\"journal\":{\"name\":\"BMC Biophysics\",\"volume\":\"5 \",\"pages\":\"2\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-01-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2046-1682-5-2\",\"citationCount\":\"23\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Biophysics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2046-1682-5-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biophysics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2046-1682-5-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Protein dynamics at Eph receptor-ligand interfaces as revealed by crystallography, NMR and MD simulations.
Background: The role of dynamics in protein functions including signal transduction is just starting to be deciphered. Eph receptors with 16 members divided into A- and B- subclasses are respectively activated by 9 A- and B-ephrin ligands. EphA4 is the only receptor capable of binding to all 9 ephrins and small molecules with overlapped interfaces.
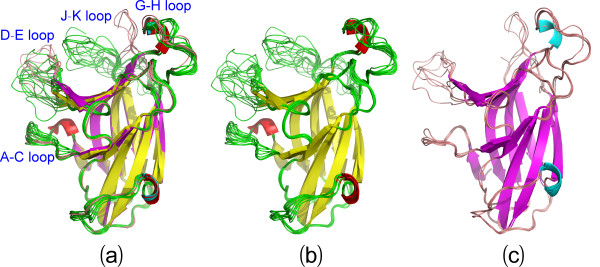
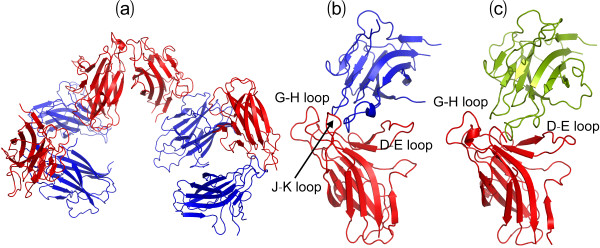
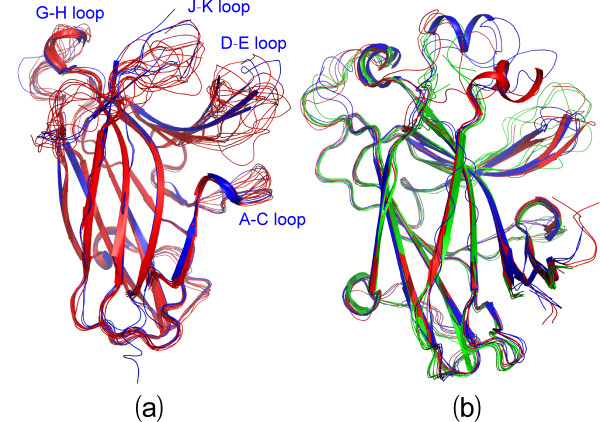
Results: We first determined the structures of the EphA4 ligand binding domain (LBD) in two crystals of P1 space group. Noticeably, 8 EphA4 molecules were found in one asymmetric unit and consequently from two crystals we obtained 16 structures, which show significant conformational variations over the functionally critical A-C, D-E, G-H and J-K loops. The 16 new structures, together with previous 9 ones, can be categorized into two groups: closed and open forms which resemble the uncomplexed and complexed structures of the EphA4 LBD respectively. To assess whether the conformational diversity over the loops primarily results from the intrinsic dynamics, we initiated 30-ns molecular dynamics (MD) simulations for both closed and open forms. The results indicate that the loops do have much higher intrinsic dynamics, which is further unravelled by NMR H/D exchange experiments. During simulations, the open form has the RMS deviations slightly larger than those of the closed one, suggesting the open form may be less stable in the absence of external contacts. Furthermore, no obvious exchange between two forms is observed within 30 ns, implying that they are dynamically separated.
Conclusions: Our study provides the first experimental and computational result revealing that the intrinsic dynamics are most likely underlying the conformational diversity observed for the EphA4 LBD loops mediating the binding affinity and specificity. Interestingly, the open conformation of the EphA4 LBD is slightly unstable in the absence of it natural ligand ephrins, implying that the conformational transition from the closed to open has to be driven by the high-affinity interaction with ephrins because the weak interaction with small molecule was found to be insufficient to trigger the transition. Our results therefore highlight the key role of protein dynamics in Eph-ephrin signalling and would benefit future design of agonists/antagonists targeting Eph receptors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
