{"title":"任意蛋白质-蛋白质对接目标是生物学相关的界面。","authors":"Juliette Martin, Richard Lavery","doi":"10.1186/2046-1682-5-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Protein-protein recognition is of fundamental importance in the vast majority of biological processes. However, it has already been demonstrated that it is very hard to distinguish true complexes from false complexes in so-called cross-docking experiments, where binary protein complexes are separated and the isolated proteins are all docked against each other and scored. Does this result, at least in part, reflect a physical reality? False complexes could reflect possible nonspecific or weak associations.</p><p><strong>Results: </strong>In this paper, we investigate the twilight zone of protein-protein interactions, building on an interesting outcome of cross-docking experiments: false complexes seem to favor residues from the true interaction site, suggesting that randomly chosen partners dock in a non-random fashion on protein surfaces. Here, we carry out arbitrary docking of a non-redundant data set of 198 proteins, with more than 300 randomly chosen \"probe\" proteins. We investigate the tendency of arbitrary partners to aggregate at localized regions of the protein surfaces, the shape and compositional bias of the generated interfaces, and the potential of this property to predict biologically relevant binding sites. We show that the non-random localization of arbitrary partners after protein-protein docking is a generic feature of protein structures. The interfaces generated in this way are not systematically planar or curved, but tend to be closer than average to the center of the proteins. These results can be used to predict biological interfaces with an AUC value up to 0.69 alone, and 0.72 when used in combination with evolutionary information. An appropriate choice of random partners and number of docking models make this method computationally practical. It is also noted that nonspecific interfaces can point to alternate interaction sites in the case of proteins with multiple interfaces. We illustrate the usefulness of arbitrary docking using PEBP (Phosphatidylethanolamine binding protein), a kinase inhibitor with multiple partners.</p><p><strong>Conclusions: </strong>An approach using arbitrary docking, and based solely on physical properties, can successfully identify biologically pertinent protein interfaces.</p>","PeriodicalId":9045,"journal":{"name":"BMC Biophysics","volume":"5 ","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2012-05-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2046-1682-5-7","citationCount":"35","resultStr":"{\"title\":\"Arbitrary protein-protein docking targets biologically relevant interfaces.\",\"authors\":\"Juliette Martin, Richard Lavery\",\"doi\":\"10.1186/2046-1682-5-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Protein-protein recognition is of fundamental importance in the vast majority of biological processes. However, it has already been demonstrated that it is very hard to distinguish true complexes from false complexes in so-called cross-docking experiments, where binary protein complexes are separated and the isolated proteins are all docked against each other and scored. Does this result, at least in part, reflect a physical reality? False complexes could reflect possible nonspecific or weak associations.</p><p><strong>Results: </strong>In this paper, we investigate the twilight zone of protein-protein interactions, building on an interesting outcome of cross-docking experiments: false complexes seem to favor residues from the true interaction site, suggesting that randomly chosen partners dock in a non-random fashion on protein surfaces. Here, we carry out arbitrary docking of a non-redundant data set of 198 proteins, with more than 300 randomly chosen \\\"probe\\\" proteins. We investigate the tendency of arbitrary partners to aggregate at localized regions of the protein surfaces, the shape and compositional bias of the generated interfaces, and the potential of this property to predict biologically relevant binding sites. We show that the non-random localization of arbitrary partners after protein-protein docking is a generic feature of protein structures. The interfaces generated in this way are not systematically planar or curved, but tend to be closer than average to the center of the proteins. These results can be used to predict biological interfaces with an AUC value up to 0.69 alone, and 0.72 when used in combination with evolutionary information. An appropriate choice of random partners and number of docking models make this method computationally practical. It is also noted that nonspecific interfaces can point to alternate interaction sites in the case of proteins with multiple interfaces. We illustrate the usefulness of arbitrary docking using PEBP (Phosphatidylethanolamine binding protein), a kinase inhibitor with multiple partners.</p><p><strong>Conclusions: </strong>An approach using arbitrary docking, and based solely on physical properties, can successfully identify biologically pertinent protein interfaces.</p>\",\"PeriodicalId\":9045,\"journal\":{\"name\":\"BMC Biophysics\",\"volume\":\"5 \",\"pages\":\"7\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-05-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2046-1682-5-7\",\"citationCount\":\"35\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Biophysics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2046-1682-5-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biophysics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2046-1682-5-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Background: Protein-protein recognition is of fundamental importance in the vast majority of biological processes. However, it has already been demonstrated that it is very hard to distinguish true complexes from false complexes in so-called cross-docking experiments, where binary protein complexes are separated and the isolated proteins are all docked against each other and scored. Does this result, at least in part, reflect a physical reality? False complexes could reflect possible nonspecific or weak associations.
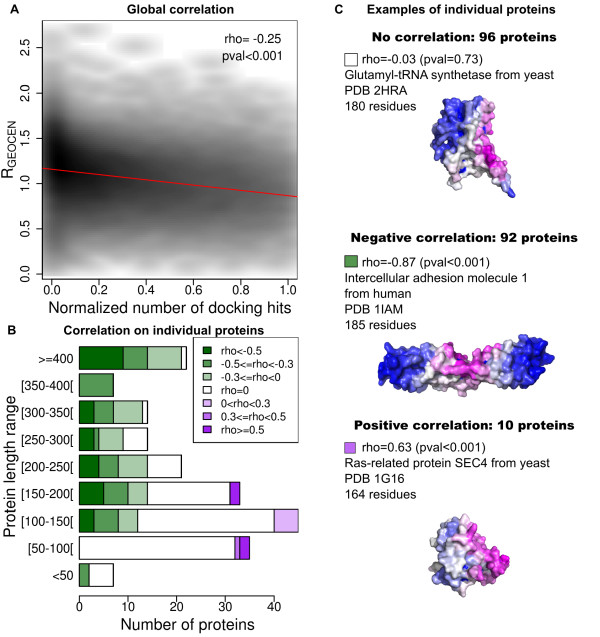
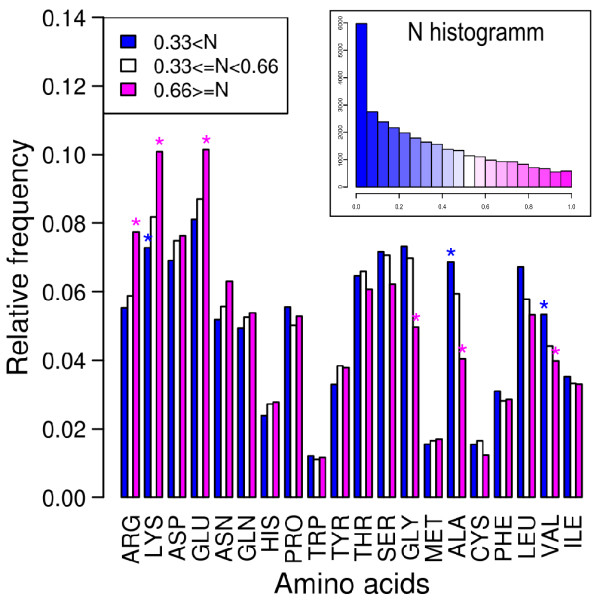
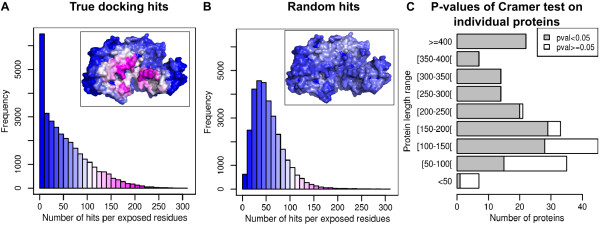
Results: In this paper, we investigate the twilight zone of protein-protein interactions, building on an interesting outcome of cross-docking experiments: false complexes seem to favor residues from the true interaction site, suggesting that randomly chosen partners dock in a non-random fashion on protein surfaces. Here, we carry out arbitrary docking of a non-redundant data set of 198 proteins, with more than 300 randomly chosen "probe" proteins. We investigate the tendency of arbitrary partners to aggregate at localized regions of the protein surfaces, the shape and compositional bias of the generated interfaces, and the potential of this property to predict biologically relevant binding sites. We show that the non-random localization of arbitrary partners after protein-protein docking is a generic feature of protein structures. The interfaces generated in this way are not systematically planar or curved, but tend to be closer than average to the center of the proteins. These results can be used to predict biological interfaces with an AUC value up to 0.69 alone, and 0.72 when used in combination with evolutionary information. An appropriate choice of random partners and number of docking models make this method computationally practical. It is also noted that nonspecific interfaces can point to alternate interaction sites in the case of proteins with multiple interfaces. We illustrate the usefulness of arbitrary docking using PEBP (Phosphatidylethanolamine binding protein), a kinase inhibitor with multiple partners.
Conclusions: An approach using arbitrary docking, and based solely on physical properties, can successfully identify biologically pertinent protein interfaces.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
