{"title":"碱基-碱基版本2:整个病毒基因组比对的单核苷酸水平分析。","authors":"William Hillary, Song-Han Lin, Chris Upton","doi":"10.1186/2042-5783-1-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Base-By-Base is a Java-based multiple sequence alignment editor. It is capable of working with protein and DNA molecules, but many of its unique features relate to the manipulation of the genomes of large DNA viruses such as poxviruses, herpesviruses, baculoviruses and asfarviruses (1-400 kb). The tool was built to serve as a platform for comparative genomics at the level of individual nucleotides.</p><p><strong>Results: </strong>In version 2, BBB-v2, of Base-By-Base we have added a series of new features aimed at providing the bench virologist with a better platform to view, annotate and analyze these complex genomes. Although a poxvirus genome, for example, may be less than 200 kb, it probably encodes close to 200 proteins using multiple classes of promoters with frequent overlapping of promoters and coding sequences and even some overlapping of genes. The new features allow users to 1) add primer annotations or other data sets in batch mode, 2) export differences between sequences to other genome browsers, 3) compare multiple genomes at a single nucleotide level of detail, 4) create new alignments from subsets/subsequences of a very large master alignment and 5) allow display of summaries of deep RNA sequencing data sets on a genome sequence.</p><p><strong>Conclusion: </strong>BBB-v2 significantly improves the ability of virologists to work with genome sequences and provides a platform with which they can use a multiple sequence alignment as the basis for their own editable documents. Also, a .bbb document, with a variety of annotations in addition to the basic coding regions, can be shared among collaborators or made available to an entire research community. The program is available via Virology.ca using Java Web Start and is platform independent; the Java 1.5 virtual machine is required.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"1 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2011-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-1-2","citationCount":"44","resultStr":"{\"title\":\"Base-By-Base version 2: single nucleotide-level analysis of whole viral genome alignments.\",\"authors\":\"William Hillary, Song-Han Lin, Chris Upton\",\"doi\":\"10.1186/2042-5783-1-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Base-By-Base is a Java-based multiple sequence alignment editor. It is capable of working with protein and DNA molecules, but many of its unique features relate to the manipulation of the genomes of large DNA viruses such as poxviruses, herpesviruses, baculoviruses and asfarviruses (1-400 kb). The tool was built to serve as a platform for comparative genomics at the level of individual nucleotides.</p><p><strong>Results: </strong>In version 2, BBB-v2, of Base-By-Base we have added a series of new features aimed at providing the bench virologist with a better platform to view, annotate and analyze these complex genomes. Although a poxvirus genome, for example, may be less than 200 kb, it probably encodes close to 200 proteins using multiple classes of promoters with frequent overlapping of promoters and coding sequences and even some overlapping of genes. The new features allow users to 1) add primer annotations or other data sets in batch mode, 2) export differences between sequences to other genome browsers, 3) compare multiple genomes at a single nucleotide level of detail, 4) create new alignments from subsets/subsequences of a very large master alignment and 5) allow display of summaries of deep RNA sequencing data sets on a genome sequence.</p><p><strong>Conclusion: </strong>BBB-v2 significantly improves the ability of virologists to work with genome sequences and provides a platform with which they can use a multiple sequence alignment as the basis for their own editable documents. Also, a .bbb document, with a variety of annotations in addition to the basic coding regions, can be shared among collaborators or made available to an entire research community. The program is available via Virology.ca using Java Web Start and is platform independent; the Java 1.5 virtual machine is required.</p>\",\"PeriodicalId\":18538,\"journal\":{\"name\":\"Microbial Informatics and Experimentation\",\"volume\":\"1 1\",\"pages\":\"2\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-06-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2042-5783-1-2\",\"citationCount\":\"44\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Informatics and Experimentation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2042-5783-1-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-1-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 44
摘要
背景:Base-By-Base是一个基于java的多序列比对编辑器。它能够处理蛋白质和DNA分子,但它的许多独特功能与操纵大型DNA病毒(如痘病毒、疱疹病毒、杆状病毒和阿斯法病毒)的基因组有关(1-400 kb)。该工具的建立是为了在单个核苷酸水平上作为比较基因组学的平台。结果:在Base-By-Base的BBB-v2版本中,我们增加了一系列新功能,旨在为实验室病毒学家提供更好的平台来查看、注释和分析这些复杂的基因组。例如,虽然痘病毒基因组可能小于200 kb,但它可能使用多种启动子编码近200种蛋白质,启动子和编码序列经常重叠,甚至一些基因重叠。新功能允许用户1)在批处理模式下添加引物注释或其他数据集,2)将序列之间的差异导出到其他基因组浏览器,3)在单个核苷酸水平上比较多个基因组的细节,4)从非常大的主序列的子集/子序列创建新的比对,5)允许显示基因组序列上的深度RNA测序数据集摘要。结论:BBB-v2显著提高了病毒学家处理基因组序列的能力,并提供了一个平台,使他们可以使用多序列比对作为他们自己编辑文档的基础。此外,除了基本编码区域之外,.bbb文档还带有各种注释,可以在合作者之间共享,或者可供整个研究社区使用。该程序可通过病毒学获得。可以使用Java Web Start并且是平台独立的;需要安装Java 1.5虚拟机。
Base-By-Base version 2: single nucleotide-level analysis of whole viral genome alignments.
Background: Base-By-Base is a Java-based multiple sequence alignment editor. It is capable of working with protein and DNA molecules, but many of its unique features relate to the manipulation of the genomes of large DNA viruses such as poxviruses, herpesviruses, baculoviruses and asfarviruses (1-400 kb). The tool was built to serve as a platform for comparative genomics at the level of individual nucleotides.
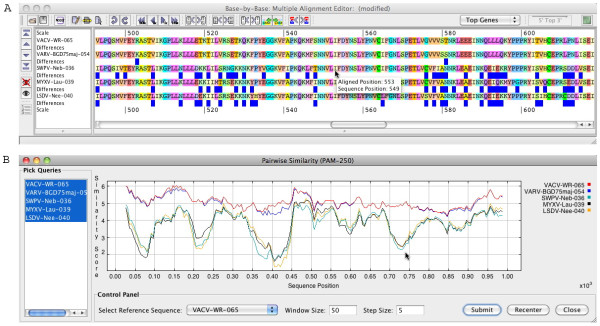
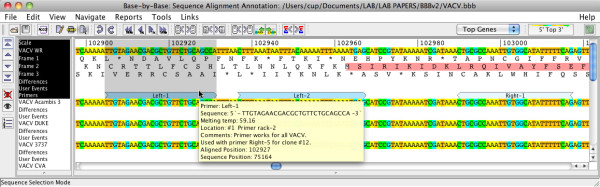
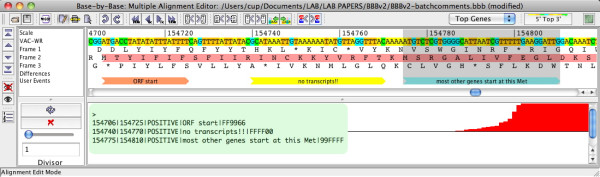
Results: In version 2, BBB-v2, of Base-By-Base we have added a series of new features aimed at providing the bench virologist with a better platform to view, annotate and analyze these complex genomes. Although a poxvirus genome, for example, may be less than 200 kb, it probably encodes close to 200 proteins using multiple classes of promoters with frequent overlapping of promoters and coding sequences and even some overlapping of genes. The new features allow users to 1) add primer annotations or other data sets in batch mode, 2) export differences between sequences to other genome browsers, 3) compare multiple genomes at a single nucleotide level of detail, 4) create new alignments from subsets/subsequences of a very large master alignment and 5) allow display of summaries of deep RNA sequencing data sets on a genome sequence.
Conclusion: BBB-v2 significantly improves the ability of virologists to work with genome sequences and provides a platform with which they can use a multiple sequence alignment as the basis for their own editable documents. Also, a .bbb document, with a variety of annotations in addition to the basic coding regions, can be shared among collaborators or made available to an entire research community. The program is available via Virology.ca using Java Web Start and is platform independent; the Java 1.5 virtual machine is required.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
