Peter E Larsen, Frank R Collart, Dawn Field, Folker Meyer, Kevin P Keegan, Christopher S Henry, John McGrath, John Quinn, Jack A Gilbert
{"title":"预测相对代谢组转换(PRMT):从沿海海洋宏基因组数据集确定代谢转换。","authors":"Peter E Larsen, Frank R Collart, Dawn Field, Folker Meyer, Kevin P Keegan, Christopher S Henry, John McGrath, John Quinn, Jack A Gilbert","doi":"10.1186/2042-5783-1-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The world's oceans are home to a diverse array of microbial life whose metabolic activity helps to drive the earth's biogeochemical cycles. Metagenomic analysis has revolutionized our access to these communities, providing a system-scale perspective of microbial community interactions. However, while metagenome sequencing can provide useful estimates of the relative change in abundance of specific genes and taxa between environments or over time, this does not investigate the relative changes in the production or consumption of different metabolites.</p><p><strong>Results: </strong>We propose a methodology, Predicted Relative Metabolic Turnover (PRMT) that defines and enables exploration of metabolite-space inferred from the metagenome. Our analysis of metagenomic data from a time-series study in the Western English Channel demonstrated considerable correlations between predicted relative metabolic turnover and seasonal changes in abundance of measured environmental parameters as well as with observed seasonal changes in bacterial population structure.</p><p><strong>Conclusions: </strong>The PRMT method was successfully applied to metagenomic data to explore the Western English Channel microbial metabalome to generate specific, biologically testable hypotheses. Generated hypotheses linked organic phosphate utilization to Gammaproteobactaria, Plantcomycetes, and Betaproteobacteria, chitin degradation to Actinomycetes, and potential small molecule biosynthesis pathways for Lentisphaerae, Chlamydiae, and Crenarchaeota. The PRMT method can be applied as a general tool for the analysis of additional metagenomic or transcriptomic datasets.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"1 1","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2011-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-1-4","citationCount":"91","resultStr":"{\"title\":\"Predicted Relative Metabolomic Turnover (PRMT): determining metabolic turnover from a coastal marine metagenomic dataset.\",\"authors\":\"Peter E Larsen, Frank R Collart, Dawn Field, Folker Meyer, Kevin P Keegan, Christopher S Henry, John McGrath, John Quinn, Jack A Gilbert\",\"doi\":\"10.1186/2042-5783-1-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The world's oceans are home to a diverse array of microbial life whose metabolic activity helps to drive the earth's biogeochemical cycles. Metagenomic analysis has revolutionized our access to these communities, providing a system-scale perspective of microbial community interactions. However, while metagenome sequencing can provide useful estimates of the relative change in abundance of specific genes and taxa between environments or over time, this does not investigate the relative changes in the production or consumption of different metabolites.</p><p><strong>Results: </strong>We propose a methodology, Predicted Relative Metabolic Turnover (PRMT) that defines and enables exploration of metabolite-space inferred from the metagenome. Our analysis of metagenomic data from a time-series study in the Western English Channel demonstrated considerable correlations between predicted relative metabolic turnover and seasonal changes in abundance of measured environmental parameters as well as with observed seasonal changes in bacterial population structure.</p><p><strong>Conclusions: </strong>The PRMT method was successfully applied to metagenomic data to explore the Western English Channel microbial metabalome to generate specific, biologically testable hypotheses. Generated hypotheses linked organic phosphate utilization to Gammaproteobactaria, Plantcomycetes, and Betaproteobacteria, chitin degradation to Actinomycetes, and potential small molecule biosynthesis pathways for Lentisphaerae, Chlamydiae, and Crenarchaeota. The PRMT method can be applied as a general tool for the analysis of additional metagenomic or transcriptomic datasets.</p>\",\"PeriodicalId\":18538,\"journal\":{\"name\":\"Microbial Informatics and Experimentation\",\"volume\":\"1 1\",\"pages\":\"4\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-06-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2042-5783-1-4\",\"citationCount\":\"91\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Informatics and Experimentation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2042-5783-1-4\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-1-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Predicted Relative Metabolomic Turnover (PRMT): determining metabolic turnover from a coastal marine metagenomic dataset.
Background: The world's oceans are home to a diverse array of microbial life whose metabolic activity helps to drive the earth's biogeochemical cycles. Metagenomic analysis has revolutionized our access to these communities, providing a system-scale perspective of microbial community interactions. However, while metagenome sequencing can provide useful estimates of the relative change in abundance of specific genes and taxa between environments or over time, this does not investigate the relative changes in the production or consumption of different metabolites.
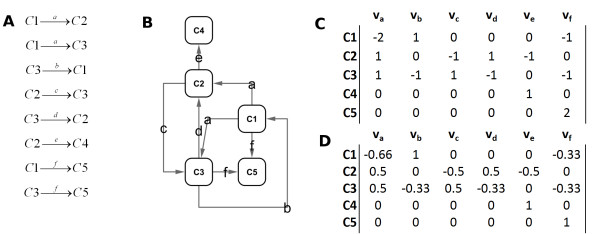
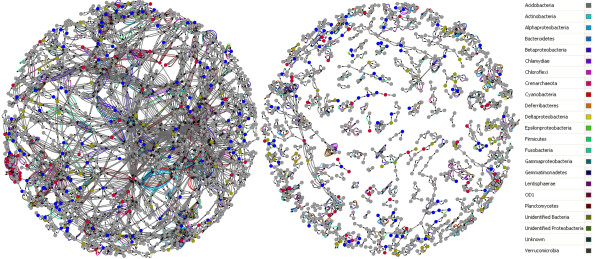
Results: We propose a methodology, Predicted Relative Metabolic Turnover (PRMT) that defines and enables exploration of metabolite-space inferred from the metagenome. Our analysis of metagenomic data from a time-series study in the Western English Channel demonstrated considerable correlations between predicted relative metabolic turnover and seasonal changes in abundance of measured environmental parameters as well as with observed seasonal changes in bacterial population structure.
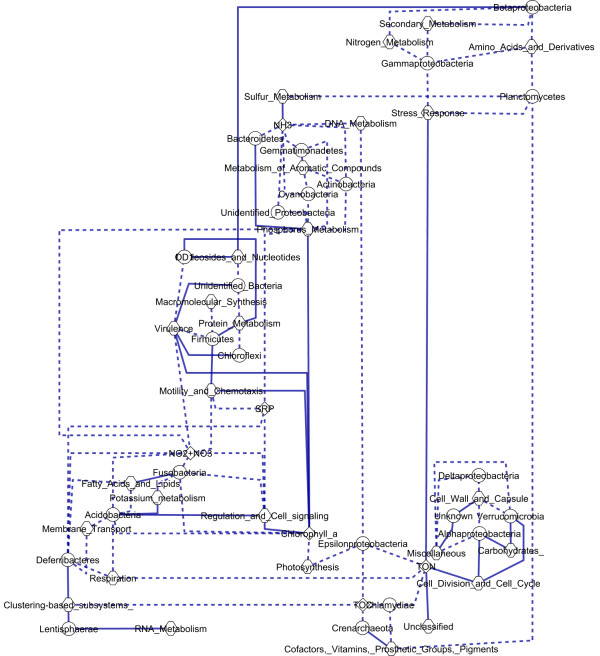
Conclusions: The PRMT method was successfully applied to metagenomic data to explore the Western English Channel microbial metabalome to generate specific, biologically testable hypotheses. Generated hypotheses linked organic phosphate utilization to Gammaproteobactaria, Plantcomycetes, and Betaproteobacteria, chitin degradation to Actinomycetes, and potential small molecule biosynthesis pathways for Lentisphaerae, Chlamydiae, and Crenarchaeota. The PRMT method can be applied as a general tool for the analysis of additional metagenomic or transcriptomic datasets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
