Seshan Ananthasubramanian, Rahul Metri, Ankur Khetan, Aman Gupta, Adam Handen, Nagasuma Chandra, Madhavi Ganapathiraju
{"title":"结核分枝杆菌和艰难梭菌相互作用组:细菌相互作用组预测计算系统快速发展的示范。","authors":"Seshan Ananthasubramanian, Rahul Metri, Ankur Khetan, Aman Gupta, Adam Handen, Nagasuma Chandra, Madhavi Ganapathiraju","doi":"10.1186/2042-5783-2-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Protein-protein interaction (PPI) networks (interactomes) of most organisms, except for some model organisms, are largely unknown. Experimental methods including high-throughput techniques are highly resource intensive. Therefore, computational discovery of PPIs can accelerate biological discovery by presenting \"most-promising\" pairs of proteins that are likely to interact. For many bacteria, genome sequence, and thereby genomic context of proteomes, is readily available; additionally, for some of these proteomes, localization and functional annotations are also available, but interactomes are not available. We present here a method for rapid development of computational system to predict interactome of bacterial proteomes. While other studies have presented methods to transfer interologs across species, here, we propose transfer of computational models to benefit from cross-species annotations, thereby predicting many more novel interactions even in the absence of interologs. Mycobacterium tuberculosis (Mtb) and Clostridium difficile (CD) have been used to demonstrate the work.</p><p><strong>Results: </strong>We developed a random forest classifier over features derived from Gene Ontology annotations and genetic context scores provided by STRING database for predicting Mtb and CD interactions independently. The Mtb classifier gave a precision of 94% and a recall of 23% on a held out test set. The Mtb model was then run on all the 8 million protein pairs of the Mtb proteome, resulting in 708 new interactions (at 94% expected precision) or 1,595 new interactions at 80% expected precision. The CD classifier gave a precision of 90% and a recall of 16% on a held out test set. The CD model was run on all the 8 million protein pairs of the CD proteome, resulting in 143 new interactions (at 90% expected precision) or 580 new interactions (at 80% expected precision). We also compared the overlap of predictions of our method with STRING database interactions for CD and Mtb and also with interactions identified recently by a bacterial 2-hybrid system for Mtb. To demonstrate the utility of transfer of computational models, we made use of the developed Mtb model and used it to predict CD protein-pairs. The cross species model thus developed yielded a precision of 88% at a recall of 8%. To demonstrate transfer of features from other organisms in the absence of feature-based and interaction-based information, we transferred missing feature values from Mtb orthologs into the CD data. In transferring this data from orthologs (not interologs), we showed that a large number of interactions can be predicted.</p><p><strong>Conclusions: </strong>Rapid discovery of (partial) bacterial interactome can be made by using existing set of GO and STRING features associated with the organisms. We can make use of cross-species interactome development, when there are not even sufficient known interactions to develop a computational prediction system. Computational model of well-studied organism(s) can be employed to make the initial interactome prediction for the target organism. We have also demonstrated successfully, that annotations can be transferred from orthologs in well-studied organisms enabling accurate predictions for organisms with no annotations. These approaches can serve as building blocks to address the challenges associated with feature coverage, missing interactions towards rapid interactome discovery for bacterial organisms.</p><p><strong>Availability: </strong>The predictions for all Mtb and CD proteins are made available at: http://severus.dbmi.pitt.edu/TB and http://severus.dbmi.pitt.edu/CD respectively for browsing as well as for download.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"2 ","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2012-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-2-4","citationCount":"10","resultStr":"{\"title\":\"Mycobacterium tuberculosis and Clostridium difficille interactomes: demonstration of rapid development of computational system for bacterial interactome prediction.\",\"authors\":\"Seshan Ananthasubramanian, Rahul Metri, Ankur Khetan, Aman Gupta, Adam Handen, Nagasuma Chandra, Madhavi Ganapathiraju\",\"doi\":\"10.1186/2042-5783-2-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Protein-protein interaction (PPI) networks (interactomes) of most organisms, except for some model organisms, are largely unknown. Experimental methods including high-throughput techniques are highly resource intensive. Therefore, computational discovery of PPIs can accelerate biological discovery by presenting \\\"most-promising\\\" pairs of proteins that are likely to interact. For many bacteria, genome sequence, and thereby genomic context of proteomes, is readily available; additionally, for some of these proteomes, localization and functional annotations are also available, but interactomes are not available. We present here a method for rapid development of computational system to predict interactome of bacterial proteomes. While other studies have presented methods to transfer interologs across species, here, we propose transfer of computational models to benefit from cross-species annotations, thereby predicting many more novel interactions even in the absence of interologs. Mycobacterium tuberculosis (Mtb) and Clostridium difficile (CD) have been used to demonstrate the work.</p><p><strong>Results: </strong>We developed a random forest classifier over features derived from Gene Ontology annotations and genetic context scores provided by STRING database for predicting Mtb and CD interactions independently. The Mtb classifier gave a precision of 94% and a recall of 23% on a held out test set. The Mtb model was then run on all the 8 million protein pairs of the Mtb proteome, resulting in 708 new interactions (at 94% expected precision) or 1,595 new interactions at 80% expected precision. The CD classifier gave a precision of 90% and a recall of 16% on a held out test set. The CD model was run on all the 8 million protein pairs of the CD proteome, resulting in 143 new interactions (at 90% expected precision) or 580 new interactions (at 80% expected precision). We also compared the overlap of predictions of our method with STRING database interactions for CD and Mtb and also with interactions identified recently by a bacterial 2-hybrid system for Mtb. To demonstrate the utility of transfer of computational models, we made use of the developed Mtb model and used it to predict CD protein-pairs. The cross species model thus developed yielded a precision of 88% at a recall of 8%. To demonstrate transfer of features from other organisms in the absence of feature-based and interaction-based information, we transferred missing feature values from Mtb orthologs into the CD data. In transferring this data from orthologs (not interologs), we showed that a large number of interactions can be predicted.</p><p><strong>Conclusions: </strong>Rapid discovery of (partial) bacterial interactome can be made by using existing set of GO and STRING features associated with the organisms. We can make use of cross-species interactome development, when there are not even sufficient known interactions to develop a computational prediction system. Computational model of well-studied organism(s) can be employed to make the initial interactome prediction for the target organism. We have also demonstrated successfully, that annotations can be transferred from orthologs in well-studied organisms enabling accurate predictions for organisms with no annotations. These approaches can serve as building blocks to address the challenges associated with feature coverage, missing interactions towards rapid interactome discovery for bacterial organisms.</p><p><strong>Availability: </strong>The predictions for all Mtb and CD proteins are made available at: http://severus.dbmi.pitt.edu/TB and http://severus.dbmi.pitt.edu/CD respectively for browsing as well as for download.</p>\",\"PeriodicalId\":18538,\"journal\":{\"name\":\"Microbial Informatics and Experimentation\",\"volume\":\"2 \",\"pages\":\"4\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2042-5783-2-4\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Microbial Informatics and Experimentation\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2042-5783-2-4\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-2-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Mycobacterium tuberculosis and Clostridium difficille interactomes: demonstration of rapid development of computational system for bacterial interactome prediction.
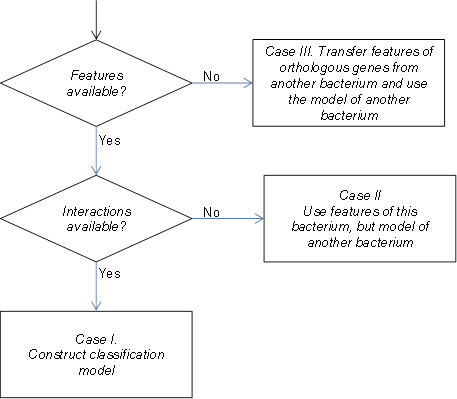
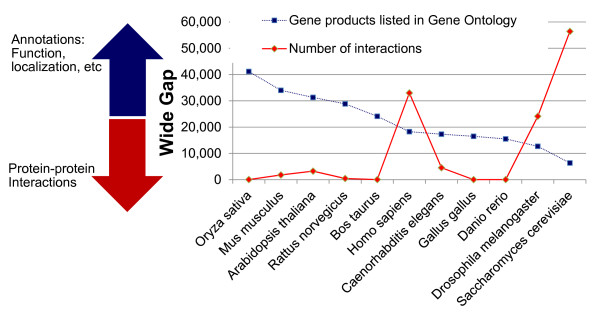
Background: Protein-protein interaction (PPI) networks (interactomes) of most organisms, except for some model organisms, are largely unknown. Experimental methods including high-throughput techniques are highly resource intensive. Therefore, computational discovery of PPIs can accelerate biological discovery by presenting "most-promising" pairs of proteins that are likely to interact. For many bacteria, genome sequence, and thereby genomic context of proteomes, is readily available; additionally, for some of these proteomes, localization and functional annotations are also available, but interactomes are not available. We present here a method for rapid development of computational system to predict interactome of bacterial proteomes. While other studies have presented methods to transfer interologs across species, here, we propose transfer of computational models to benefit from cross-species annotations, thereby predicting many more novel interactions even in the absence of interologs. Mycobacterium tuberculosis (Mtb) and Clostridium difficile (CD) have been used to demonstrate the work.
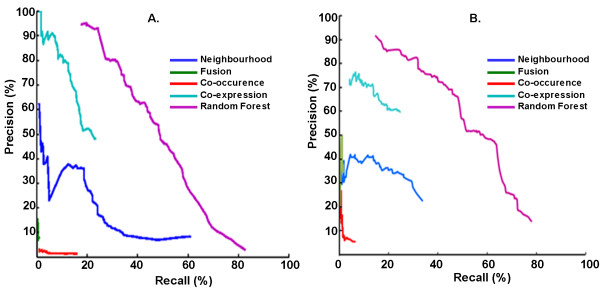
Results: We developed a random forest classifier over features derived from Gene Ontology annotations and genetic context scores provided by STRING database for predicting Mtb and CD interactions independently. The Mtb classifier gave a precision of 94% and a recall of 23% on a held out test set. The Mtb model was then run on all the 8 million protein pairs of the Mtb proteome, resulting in 708 new interactions (at 94% expected precision) or 1,595 new interactions at 80% expected precision. The CD classifier gave a precision of 90% and a recall of 16% on a held out test set. The CD model was run on all the 8 million protein pairs of the CD proteome, resulting in 143 new interactions (at 90% expected precision) or 580 new interactions (at 80% expected precision). We also compared the overlap of predictions of our method with STRING database interactions for CD and Mtb and also with interactions identified recently by a bacterial 2-hybrid system for Mtb. To demonstrate the utility of transfer of computational models, we made use of the developed Mtb model and used it to predict CD protein-pairs. The cross species model thus developed yielded a precision of 88% at a recall of 8%. To demonstrate transfer of features from other organisms in the absence of feature-based and interaction-based information, we transferred missing feature values from Mtb orthologs into the CD data. In transferring this data from orthologs (not interologs), we showed that a large number of interactions can be predicted.
Conclusions: Rapid discovery of (partial) bacterial interactome can be made by using existing set of GO and STRING features associated with the organisms. We can make use of cross-species interactome development, when there are not even sufficient known interactions to develop a computational prediction system. Computational model of well-studied organism(s) can be employed to make the initial interactome prediction for the target organism. We have also demonstrated successfully, that annotations can be transferred from orthologs in well-studied organisms enabling accurate predictions for organisms with no annotations. These approaches can serve as building blocks to address the challenges associated with feature coverage, missing interactions towards rapid interactome discovery for bacterial organisms.
Availability: The predictions for all Mtb and CD proteins are made available at: http://severus.dbmi.pitt.edu/TB and http://severus.dbmi.pitt.edu/CD respectively for browsing as well as for download.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
