Xiaowei Guan, Mark R Chance, Jill S Barnholtz-Sloan
{"title":"分裂随机森林(SRF)用于确定区分癌症亚型的紧凑基因集。","authors":"Xiaowei Guan, Mark R Chance, Jill S Barnholtz-Sloan","doi":"10.1186/2043-9113-2-13","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The identification of very small subsets of predictive variables is an important toπc that has not often been considered in the literature. In order to discover highly predictive yet compact gene set classifiers from whole genome expression data, a non-parametric, iterative algorithm, Splitting Random Forest (SRF), was developed to robustly identify genes that distinguish between molecular subtypes. The goal is to improve the prediction accuracy while considering sparsity.</p><p><strong>Results: </strong>The optimal SRF 50 run (SRF50) gene classifiers for glioblastoma (GB), breast (BC) and ovarian cancer (OC) subtypes had overall prediction rates comparable to those from published datasets upon validation (80.1%-91.7%). The SRF50 sets outperformed other methods by identifying compact gene sets needed for distinguishing between tested cancer subtypes (10-200 fold fewer genes than ANOVA or published gene sets). The SRF50 sets achieved superior and robust overall and subtype prediction accuracies when compared with single random forest (RF) and the Top 50 ANOVA results (80.1% vs 77.8% for GB; 84.0% vs 74.1% for BC; 89.8% vs 88.9% for OC in SRF50 vs single RF comparison; 80.1% vs 77.2% for GB; 84.0% vs 82.7% for BC; 89.8% vs 87.0% for OC in SRF50 vs Top 50 ANOVA comparison). There was significant overlap between SRF50 and published gene sets, showing that SRF identifies the relevant sub-sets of important gene lists. Through Ingenuity Pathway Analysis (IPA), the overlap in \"hub\" genes between the SRF50 and published genes sets were RB1, πK3R1, PDGFBB and ERK1/2 for GB; ESR1, MYC, NFkB and ERK1/2 for BC; and Akt, FN1, NFkB, PDGFBB and ERK1/2 for OC.</p><p><strong>Conclusions: </strong>The SRF approach is an effective driver of biomarker discovery research that reduces the number of genes needed for robust classification, dissects complex, high dimensional \"omic\" data and provides novel insights into the cellular mechanisms that define cancer subtypes.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"2 1","pages":"13"},"PeriodicalIF":0.0000,"publicationDate":"2012-05-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-2-13","citationCount":"44","resultStr":"{\"title\":\"Splitting random forest (SRF) for determining compact sets of genes that distinguish between cancer subtypes.\",\"authors\":\"Xiaowei Guan, Mark R Chance, Jill S Barnholtz-Sloan\",\"doi\":\"10.1186/2043-9113-2-13\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The identification of very small subsets of predictive variables is an important toπc that has not often been considered in the literature. In order to discover highly predictive yet compact gene set classifiers from whole genome expression data, a non-parametric, iterative algorithm, Splitting Random Forest (SRF), was developed to robustly identify genes that distinguish between molecular subtypes. The goal is to improve the prediction accuracy while considering sparsity.</p><p><strong>Results: </strong>The optimal SRF 50 run (SRF50) gene classifiers for glioblastoma (GB), breast (BC) and ovarian cancer (OC) subtypes had overall prediction rates comparable to those from published datasets upon validation (80.1%-91.7%). The SRF50 sets outperformed other methods by identifying compact gene sets needed for distinguishing between tested cancer subtypes (10-200 fold fewer genes than ANOVA or published gene sets). The SRF50 sets achieved superior and robust overall and subtype prediction accuracies when compared with single random forest (RF) and the Top 50 ANOVA results (80.1% vs 77.8% for GB; 84.0% vs 74.1% for BC; 89.8% vs 88.9% for OC in SRF50 vs single RF comparison; 80.1% vs 77.2% for GB; 84.0% vs 82.7% for BC; 89.8% vs 87.0% for OC in SRF50 vs Top 50 ANOVA comparison). There was significant overlap between SRF50 and published gene sets, showing that SRF identifies the relevant sub-sets of important gene lists. Through Ingenuity Pathway Analysis (IPA), the overlap in \\\"hub\\\" genes between the SRF50 and published genes sets were RB1, πK3R1, PDGFBB and ERK1/2 for GB; ESR1, MYC, NFkB and ERK1/2 for BC; and Akt, FN1, NFkB, PDGFBB and ERK1/2 for OC.</p><p><strong>Conclusions: </strong>The SRF approach is an effective driver of biomarker discovery research that reduces the number of genes needed for robust classification, dissects complex, high dimensional \\\"omic\\\" data and provides novel insights into the cellular mechanisms that define cancer subtypes.</p>\",\"PeriodicalId\":73663,\"journal\":{\"name\":\"Journal of clinical bioinformatics\",\"volume\":\"2 1\",\"pages\":\"13\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-05-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2043-9113-2-13\",\"citationCount\":\"44\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2043-9113-2-13\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-2-13","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 44
摘要
背景:识别非常小的预测变量子集是一个重要的问题,但在文献中很少被考虑到。为了从全基因组表达数据中发现高度预测且紧凑的基因集分类器,开发了一种非参数迭代算法——分裂随机森林(SRF),以稳健地识别区分分子亚型的基因。目标是在考虑稀疏性的同时提高预测精度。结果:胶质母细胞瘤(GB)、乳腺癌(BC)和卵巢癌(OC)亚型的最佳SRF50基因分类器(SRF50)在验证时的总体预测率与已发表的数据集相当(80.1%-91.7%)。SRF50组通过识别用于区分被测试癌症亚型所需的紧凑基因集(比方差分析或已发表的基因集少10-200倍)优于其他方法。与单一随机森林(RF)和前50名ANOVA结果相比,SRF50集的总体和亚型预测精度更高,更稳健(80.1% vs 77.8%;84.0% vs 74.1% BC;SRF50和单一RF对比中OC的比例分别为89.8%和88.9%;80.1% vs GB 77.2%;84.0% vs BC 82.7%;在SRF50和前50名的方差分析比较中,OC的比例为89.8%和87.0%)。SRF50与已发表的基因集之间存在显著的重叠,表明SRF识别了重要基因列表的相关亚集。通过匠心途径分析(Ingenuity Pathway Analysis, IPA), SRF50与已发表基因集的“枢纽”基因重叠为GB的RB1、πK3R1、PDGFBB和ERK1/2;BC的ESR1、MYC、NFkB和ERK1/2;Akt、FN1、NFkB、PDGFBB和ERK1/2。结论:SRF方法是生物标志物发现研究的有效驱动因素,减少了强大分类所需的基因数量,剖析了复杂的高维“组学”数据,并为定义癌症亚型的细胞机制提供了新的见解。
Splitting random forest (SRF) for determining compact sets of genes that distinguish between cancer subtypes.
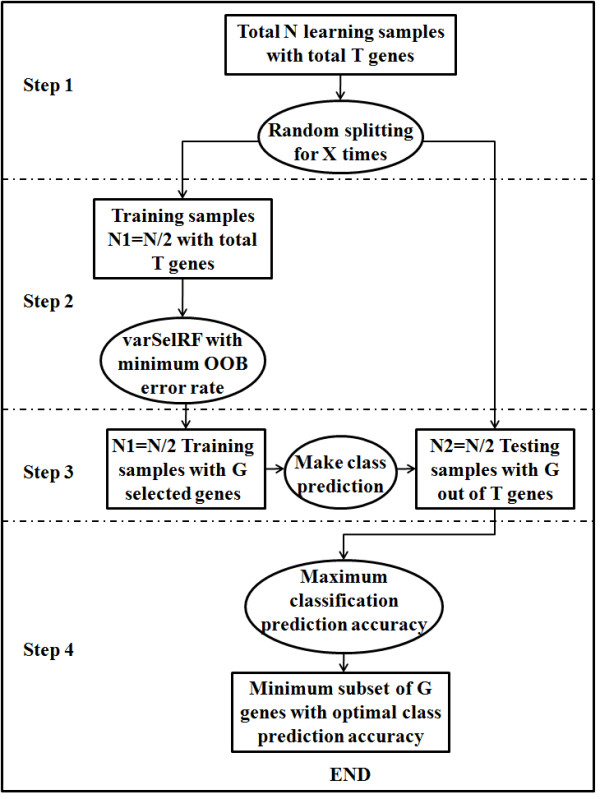
Background: The identification of very small subsets of predictive variables is an important toπc that has not often been considered in the literature. In order to discover highly predictive yet compact gene set classifiers from whole genome expression data, a non-parametric, iterative algorithm, Splitting Random Forest (SRF), was developed to robustly identify genes that distinguish between molecular subtypes. The goal is to improve the prediction accuracy while considering sparsity.
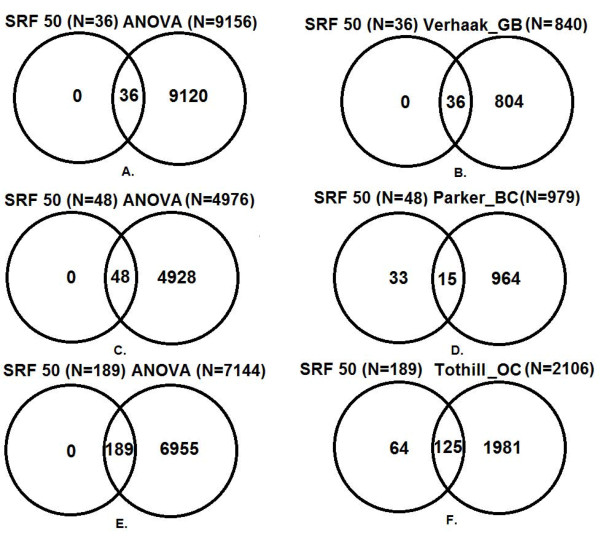
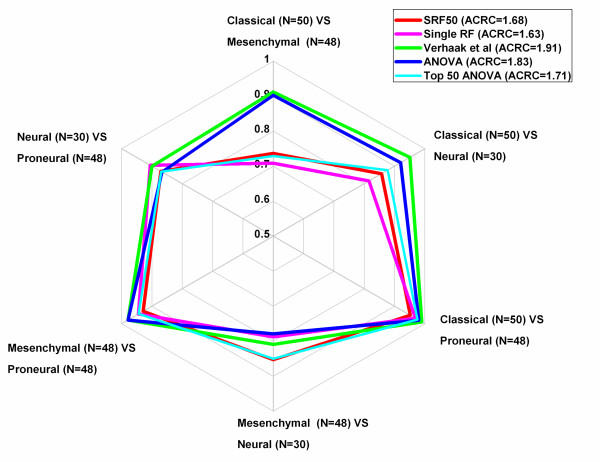
Results: The optimal SRF 50 run (SRF50) gene classifiers for glioblastoma (GB), breast (BC) and ovarian cancer (OC) subtypes had overall prediction rates comparable to those from published datasets upon validation (80.1%-91.7%). The SRF50 sets outperformed other methods by identifying compact gene sets needed for distinguishing between tested cancer subtypes (10-200 fold fewer genes than ANOVA or published gene sets). The SRF50 sets achieved superior and robust overall and subtype prediction accuracies when compared with single random forest (RF) and the Top 50 ANOVA results (80.1% vs 77.8% for GB; 84.0% vs 74.1% for BC; 89.8% vs 88.9% for OC in SRF50 vs single RF comparison; 80.1% vs 77.2% for GB; 84.0% vs 82.7% for BC; 89.8% vs 87.0% for OC in SRF50 vs Top 50 ANOVA comparison). There was significant overlap between SRF50 and published gene sets, showing that SRF identifies the relevant sub-sets of important gene lists. Through Ingenuity Pathway Analysis (IPA), the overlap in "hub" genes between the SRF50 and published genes sets were RB1, πK3R1, PDGFBB and ERK1/2 for GB; ESR1, MYC, NFkB and ERK1/2 for BC; and Akt, FN1, NFkB, PDGFBB and ERK1/2 for OC.
Conclusions: The SRF approach is an effective driver of biomarker discovery research that reduces the number of genes needed for robust classification, dissects complex, high dimensional "omic" data and provides novel insights into the cellular mechanisms that define cancer subtypes.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
