Kirk K Durston, David Ky Chiu, Andrew Kc Wong, Gary Cl Li
{"title":"亚分子分层蛋白质结构中位点相互依赖关系的统计发现。","authors":"Kirk K Durston, David Ky Chiu, Andrew Kc Wong, Gary Cl Li","doi":"10.1186/1687-4153-2012-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Much progress has been made in understanding the 3D structure of proteins using methods such as NMR and X-ray crystallography. The resulting 3D structures are extremely informative, but do not always reveal which sites and residues within the structure are of special importance. Recently, there are indications that multiple-residue, sub-domain structural relationships within the larger 3D consensus structure of a protein can be inferred from the analysis of the multiple sequence alignment data of a protein family. These intra-dependent clusters of associated sites are used to indicate hierarchical inter-residue relationships within the 3D structure. To reveal the patterns of associations among individual amino acids or sub-domain components within the structure, we apply a k-modes attribute (aligned site) clustering algorithm to the ubiquitin and transthyretin families in order to discover associations among groups of sites within the multiple sequence alignment. We then observe what these associations imply within the 3D structure of these two protein families.</p><p><strong>Results: </strong>The k-modes site clustering algorithm we developed maximizes the intra-group interdependencies based on a normalized mutual information measure. The clusters formed correspond to sub-structural components or binding and interface locations. Applying this data-directed method to the ubiquitin and transthyretin protein family multiple sequence alignments as a test bed, we located numerous interesting associations of interdependent sites. These clusters were then arranged into cluster tree diagrams which revealed four structural sub-domains within the single domain structure of ubiquitin and a single large sub-domain within transthyretin associated with the interface among transthyretin monomers. In addition, several clusters of mutually interdependent sites were discovered for each protein family, each of which appear to play an important role in the molecular structure and/or function.</p><p><strong>Conclusions: </strong>Our results demonstrate that the method we present here using a k-modes site clustering algorithm based on interdependency evaluation among sites obtained from a sequence alignment of homologous proteins can provide significant insights into the complex, hierarchical inter-residue structural relationships within the 3D structure of a protein family.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2012 1","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2012-07-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3524763/pdf/","citationCount":"0","resultStr":"{\"title\":\"Statistical discovery of site inter-dependencies in sub-molecular hierarchical protein structuring.\",\"authors\":\"Kirk K Durston, David Ky Chiu, Andrew Kc Wong, Gary Cl Li\",\"doi\":\"10.1186/1687-4153-2012-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Much progress has been made in understanding the 3D structure of proteins using methods such as NMR and X-ray crystallography. The resulting 3D structures are extremely informative, but do not always reveal which sites and residues within the structure are of special importance. Recently, there are indications that multiple-residue, sub-domain structural relationships within the larger 3D consensus structure of a protein can be inferred from the analysis of the multiple sequence alignment data of a protein family. These intra-dependent clusters of associated sites are used to indicate hierarchical inter-residue relationships within the 3D structure. To reveal the patterns of associations among individual amino acids or sub-domain components within the structure, we apply a k-modes attribute (aligned site) clustering algorithm to the ubiquitin and transthyretin families in order to discover associations among groups of sites within the multiple sequence alignment. We then observe what these associations imply within the 3D structure of these two protein families.</p><p><strong>Results: </strong>The k-modes site clustering algorithm we developed maximizes the intra-group interdependencies based on a normalized mutual information measure. The clusters formed correspond to sub-structural components or binding and interface locations. Applying this data-directed method to the ubiquitin and transthyretin protein family multiple sequence alignments as a test bed, we located numerous interesting associations of interdependent sites. These clusters were then arranged into cluster tree diagrams which revealed four structural sub-domains within the single domain structure of ubiquitin and a single large sub-domain within transthyretin associated with the interface among transthyretin monomers. In addition, several clusters of mutually interdependent sites were discovered for each protein family, each of which appear to play an important role in the molecular structure and/or function.</p><p><strong>Conclusions: </strong>Our results demonstrate that the method we present here using a k-modes site clustering algorithm based on interdependency evaluation among sites obtained from a sequence alignment of homologous proteins can provide significant insights into the complex, hierarchical inter-residue structural relationships within the 3D structure of a protein family.</p>\",\"PeriodicalId\":72957,\"journal\":{\"name\":\"EURASIP journal on bioinformatics & systems biology\",\"volume\":\"2012 1\",\"pages\":\"8\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-07-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3524763/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EURASIP journal on bioinformatics & systems biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1687-4153-2012-8\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2012-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
无标签:背景:利用核磁共振和 X 射线晶体学等方法了解蛋白质的三维结构已经取得了很大进展。由此产生的三维结构信息量极大,但并不总能揭示结构中哪些位点和残基特别重要。最近,有迹象表明,通过分析蛋白质家族的多序列比对数据,可以推断出蛋白质较大三维共识结构中的多残基、亚域结构关系。这些相关位点的内部依赖性群组被用来表示三维结构中的层次性残基间关系。为了揭示结构中单个氨基酸或子域成分之间的关联模式,我们在泛素和转hyretin家族中应用了k-modes属性(对齐位点)聚类算法,以发现多序列比对中各组位点之间的关联。然后,我们观察这些关联在这两个蛋白质家族的三维结构中意味着什么:我们开发的 k 模式位点聚类算法根据归一化互信息度量最大化了组内相互依存关系。所形成的聚类与亚结构成分或结合和界面位置相对应。以泛素和转甲状腺素蛋白家族的多序列排列为试验平台,应用这种以数据为导向的方法,我们找到了许多有趣的相互依赖位点关联。然后将这些聚类排列成聚类树图,发现泛素单结构域中有四个结构子域,而转酪蛋白中有一个大的子域与转酪蛋白单体之间的界面有关。此外,每个蛋白质家族都发现了几个相互依存的位点群,每个位点群似乎都在分子结构和/或功能中发挥着重要作用:我们的研究结果表明,我们在此介绍的基于同源蛋白质序列比对中获得的位点间相互依存性评估的 k-模式位点聚类算法,可以为深入了解蛋白质家族三维结构中复杂的、分层的残基间结构关系提供重要信息。
Statistical discovery of site inter-dependencies in sub-molecular hierarchical protein structuring.
Unlabelled:
Background: Much progress has been made in understanding the 3D structure of proteins using methods such as NMR and X-ray crystallography. The resulting 3D structures are extremely informative, but do not always reveal which sites and residues within the structure are of special importance. Recently, there are indications that multiple-residue, sub-domain structural relationships within the larger 3D consensus structure of a protein can be inferred from the analysis of the multiple sequence alignment data of a protein family. These intra-dependent clusters of associated sites are used to indicate hierarchical inter-residue relationships within the 3D structure. To reveal the patterns of associations among individual amino acids or sub-domain components within the structure, we apply a k-modes attribute (aligned site) clustering algorithm to the ubiquitin and transthyretin families in order to discover associations among groups of sites within the multiple sequence alignment. We then observe what these associations imply within the 3D structure of these two protein families.
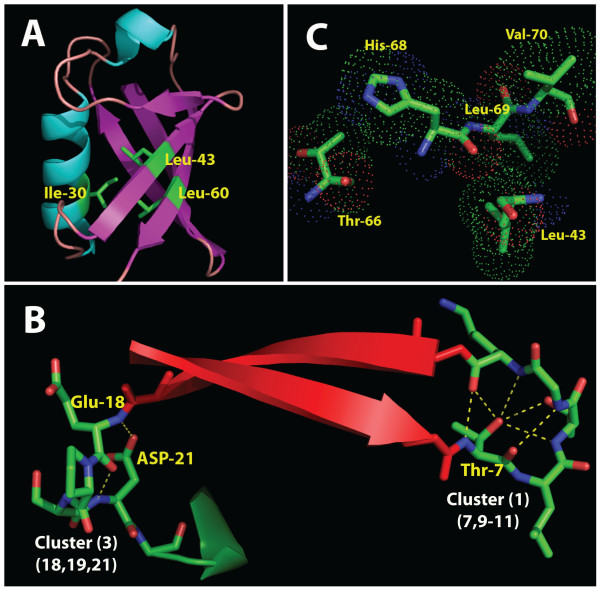
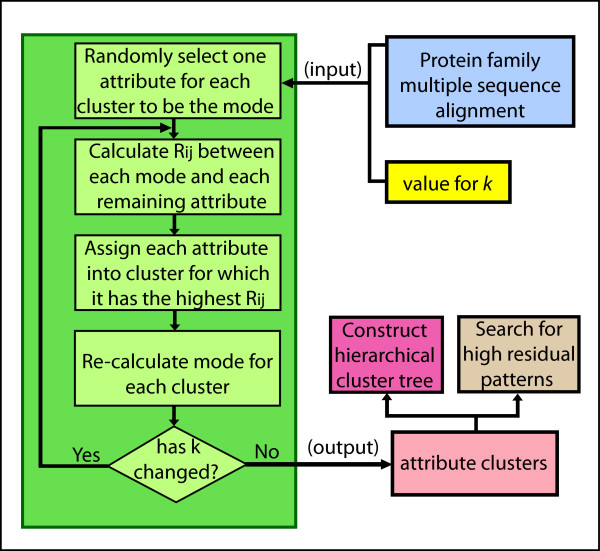
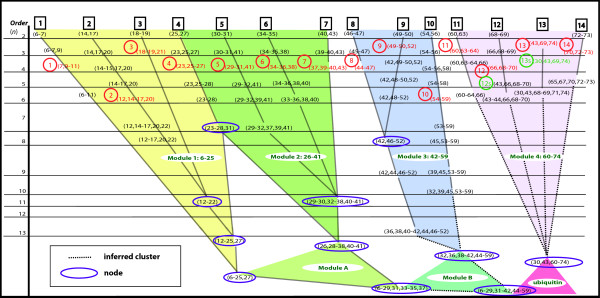
Results: The k-modes site clustering algorithm we developed maximizes the intra-group interdependencies based on a normalized mutual information measure. The clusters formed correspond to sub-structural components or binding and interface locations. Applying this data-directed method to the ubiquitin and transthyretin protein family multiple sequence alignments as a test bed, we located numerous interesting associations of interdependent sites. These clusters were then arranged into cluster tree diagrams which revealed four structural sub-domains within the single domain structure of ubiquitin and a single large sub-domain within transthyretin associated with the interface among transthyretin monomers. In addition, several clusters of mutually interdependent sites were discovered for each protein family, each of which appear to play an important role in the molecular structure and/or function.
Conclusions: Our results demonstrate that the method we present here using a k-modes site clustering algorithm based on interdependency evaluation among sites obtained from a sequence alignment of homologous proteins can provide significant insights into the complex, hierarchical inter-residue structural relationships within the 3D structure of a protein family.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
