{"title":"串联质谱的高质量精度磷酸肽鉴定。","authors":"Rovshan G Sadygov","doi":"10.1155/2012/104681","DOIUrl":null,"url":null,"abstract":"<p><p>Phosphoproteomics is a powerful analytical platform for identification and quantification of phosphorylated peptides and assignment of phosphorylation sites. Bioinformatics tools to identify phosphorylated peptides from their tandem mass spectra and protein sequence databases are important part of phosphoproteomics. In this work, we discuss general informatics aspects of mass-spectrometry-based phosphoproteomics. Some of the specifics of phosphopeptide identifications stem from the labile nature of phosphor groups and expanded peptide search space. Allowing for modifications of Ser, Thr, and Tyr residues exponentially increases effective database size. High mass resolution and accuracy measurements of precursor mass-to-charge ratios help to restrict the search space of candidate peptide sequences. The higher-order fragmentations of neutral loss ions enhance the fragment ion mass spectra of phosphorylated peptides. We show an example of a phosphopeptide identification where accounting for fragmentation from neutral loss species improves the identification scores in a database search algorithm by 50%.</p>","PeriodicalId":73474,"journal":{"name":"International journal of proteomics","volume":"2012 ","pages":"104681"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2012/104681","citationCount":"2","resultStr":"{\"title\":\"High mass accuracy phosphopeptide identification using tandem mass spectra.\",\"authors\":\"Rovshan G Sadygov\",\"doi\":\"10.1155/2012/104681\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Phosphoproteomics is a powerful analytical platform for identification and quantification of phosphorylated peptides and assignment of phosphorylation sites. Bioinformatics tools to identify phosphorylated peptides from their tandem mass spectra and protein sequence databases are important part of phosphoproteomics. In this work, we discuss general informatics aspects of mass-spectrometry-based phosphoproteomics. Some of the specifics of phosphopeptide identifications stem from the labile nature of phosphor groups and expanded peptide search space. Allowing for modifications of Ser, Thr, and Tyr residues exponentially increases effective database size. High mass resolution and accuracy measurements of precursor mass-to-charge ratios help to restrict the search space of candidate peptide sequences. The higher-order fragmentations of neutral loss ions enhance the fragment ion mass spectra of phosphorylated peptides. We show an example of a phosphopeptide identification where accounting for fragmentation from neutral loss species improves the identification scores in a database search algorithm by 50%.</p>\",\"PeriodicalId\":73474,\"journal\":{\"name\":\"International journal of proteomics\",\"volume\":\"2012 \",\"pages\":\"104681\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2012/104681\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International journal of proteomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2012/104681\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2012/7/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2012/104681","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/7/15 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
High mass accuracy phosphopeptide identification using tandem mass spectra.
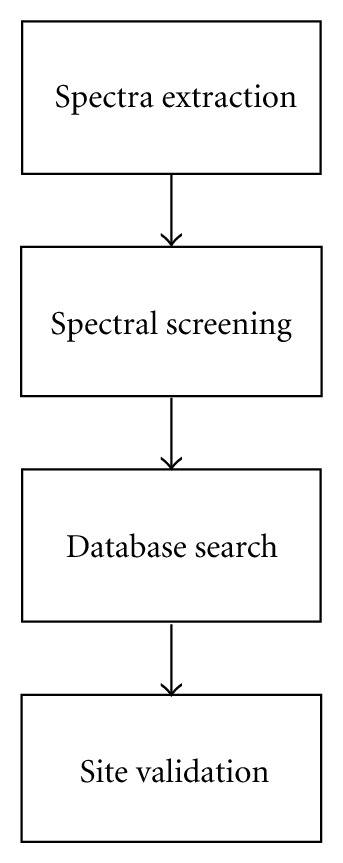
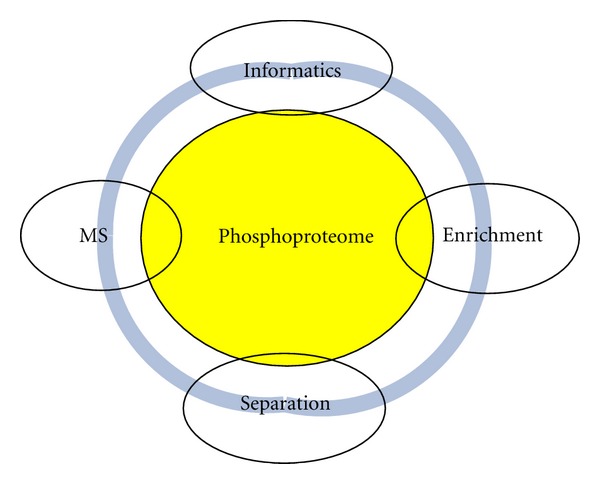
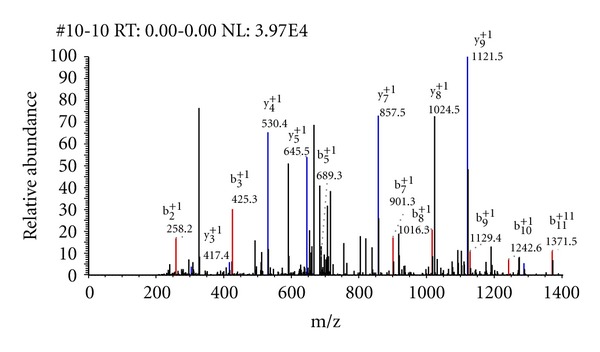
Phosphoproteomics is a powerful analytical platform for identification and quantification of phosphorylated peptides and assignment of phosphorylation sites. Bioinformatics tools to identify phosphorylated peptides from their tandem mass spectra and protein sequence databases are important part of phosphoproteomics. In this work, we discuss general informatics aspects of mass-spectrometry-based phosphoproteomics. Some of the specifics of phosphopeptide identifications stem from the labile nature of phosphor groups and expanded peptide search space. Allowing for modifications of Ser, Thr, and Tyr residues exponentially increases effective database size. High mass resolution and accuracy measurements of precursor mass-to-charge ratios help to restrict the search space of candidate peptide sequences. The higher-order fragmentations of neutral loss ions enhance the fragment ion mass spectra of phosphorylated peptides. We show an example of a phosphopeptide identification where accounting for fragmentation from neutral loss species improves the identification scores in a database search algorithm by 50%.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
