Camilo Valdes, Pearl Seo, Nicholas Tsinoremas, Jennifer Clarke
{"title":"利用RNA-Seq和微阵列技术研究伪异种移植标本的交叉杂交和交叉比对表达特征。","authors":"Camilo Valdes, Pearl Seo, Nicholas Tsinoremas, Jennifer Clarke","doi":"10.1186/2043-9113-3-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Exploring stromal changes associated with tumor growth and development is a growing area of oncologic research. In order to study molecular changes in the stroma it is recommended to separate tumor tissue from stromal tissue. This is relevant to xenograft models where tumors can be small and difficult to separate from host tissue. We introduce a novel definition of cross-alignment/cross-hybridization to compare qualitatively the ability of high-throughput mRNA sequencing, RNA-Seq, and microarrays to detect tumor and stromal expression from mixed 'pseudo-xenograft' samples vis-à-vis genes and pathways in cross-alignment (RNA-Seq) and cross-hybridization (microarrays). Samples consisted of normal mouse lung and human breast cancer cells; these were combined in fixed proportions to create a titration series of 25% steps. Our definition identifies genes in a given species (human or mouse) with undetectable expression in same-species RNA but detectable expression in cross-species RNA. We demonstrate the comparative value of this method and discuss its potential contribution in cancer research.</p><p><strong>Results: </strong>Our method can identify genes from either species that demonstrate cross-hybridization and/or cross-alignment properties. Surprisingly, the set of genes identified using a simpler and more common approach (using a 'pure' cross-species sample and calling all detected genes as 'crossers') is not a superset of the genes identified using our technique. The observed levels of cross-hybridization are relatively low: 5.3% of human genes detected in mouse, and 3.5% of mouse genes detected in human. Observed levels of cross-alignment are practically comparable to the levels of cross-hybridization: 6.5% of human genes detected in mouse, and 2.3% of mouse genes detected in human. We also observed a relatively high percentage of orthologs: 40.3% of cross-hybridizing genes, and 32.2% of cross-aligning genes.Normalizing the gene catalog to use Consensus Coding Sequence (CCDS) IDs (Genome Res 19:1316-1323, 2009), our results show that the observed levels of cross-hybridization are low: 2.7% of human CCDS IDs are detected in mouse, and 2.4% of mouse CCDS IDs are detected in human. Levels of cross-alignment using the RNA-Seq data are comparable for the mouse, 2.2% of mouse CCDS IDs detected in human, and 9.9% of human CCDS IDs detected in mouse. However, the lists of cross-aligning/cross-hybridizing genes contain many that are of specific interest to oncologic researchers.</p><p><strong>Conclusions: </strong>The conservative definition that we propose identifies genes in mouse whose expression can be attributed to human RNA, and vice versa, as well as revealing genes with cross-alignment/cross-hybridization properties which could not be identified using a simpler but more established approach. The overall percentage of genes affected by cross-hybridization/cross-alignment is small, but includes genes that are of interest to oncologic researchers. Which platform to use with mixed xenograft samples, microarrays or RNA-Seq, appears to be primarily a question of cost and whether the detection and measurement of expression of specific genes of interest are likely to be affected by cross-hybridization or cross-alignment.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":" ","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2013-04-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-3-8","citationCount":"6","resultStr":"{\"title\":\"Characteristics of cross-hybridization and cross-alignment of expression in pseudo-xenograft samples by RNA-Seq and microarrays.\",\"authors\":\"Camilo Valdes, Pearl Seo, Nicholas Tsinoremas, Jennifer Clarke\",\"doi\":\"10.1186/2043-9113-3-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Exploring stromal changes associated with tumor growth and development is a growing area of oncologic research. In order to study molecular changes in the stroma it is recommended to separate tumor tissue from stromal tissue. This is relevant to xenograft models where tumors can be small and difficult to separate from host tissue. We introduce a novel definition of cross-alignment/cross-hybridization to compare qualitatively the ability of high-throughput mRNA sequencing, RNA-Seq, and microarrays to detect tumor and stromal expression from mixed 'pseudo-xenograft' samples vis-à-vis genes and pathways in cross-alignment (RNA-Seq) and cross-hybridization (microarrays). Samples consisted of normal mouse lung and human breast cancer cells; these were combined in fixed proportions to create a titration series of 25% steps. Our definition identifies genes in a given species (human or mouse) with undetectable expression in same-species RNA but detectable expression in cross-species RNA. We demonstrate the comparative value of this method and discuss its potential contribution in cancer research.</p><p><strong>Results: </strong>Our method can identify genes from either species that demonstrate cross-hybridization and/or cross-alignment properties. Surprisingly, the set of genes identified using a simpler and more common approach (using a 'pure' cross-species sample and calling all detected genes as 'crossers') is not a superset of the genes identified using our technique. The observed levels of cross-hybridization are relatively low: 5.3% of human genes detected in mouse, and 3.5% of mouse genes detected in human. Observed levels of cross-alignment are practically comparable to the levels of cross-hybridization: 6.5% of human genes detected in mouse, and 2.3% of mouse genes detected in human. We also observed a relatively high percentage of orthologs: 40.3% of cross-hybridizing genes, and 32.2% of cross-aligning genes.Normalizing the gene catalog to use Consensus Coding Sequence (CCDS) IDs (Genome Res 19:1316-1323, 2009), our results show that the observed levels of cross-hybridization are low: 2.7% of human CCDS IDs are detected in mouse, and 2.4% of mouse CCDS IDs are detected in human. Levels of cross-alignment using the RNA-Seq data are comparable for the mouse, 2.2% of mouse CCDS IDs detected in human, and 9.9% of human CCDS IDs detected in mouse. However, the lists of cross-aligning/cross-hybridizing genes contain many that are of specific interest to oncologic researchers.</p><p><strong>Conclusions: </strong>The conservative definition that we propose identifies genes in mouse whose expression can be attributed to human RNA, and vice versa, as well as revealing genes with cross-alignment/cross-hybridization properties which could not be identified using a simpler but more established approach. The overall percentage of genes affected by cross-hybridization/cross-alignment is small, but includes genes that are of interest to oncologic researchers. Which platform to use with mixed xenograft samples, microarrays or RNA-Seq, appears to be primarily a question of cost and whether the detection and measurement of expression of specific genes of interest are likely to be affected by cross-hybridization or cross-alignment.</p>\",\"PeriodicalId\":73663,\"journal\":{\"name\":\"Journal of clinical bioinformatics\",\"volume\":\" \",\"pages\":\"8\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-04-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2043-9113-3-8\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2043-9113-3-8\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-3-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
摘要
背景:探索与肿瘤生长和发展相关的间质变化是肿瘤学研究的一个新兴领域。为了研究基质的分子变化,建议将肿瘤组织与基质组织分离。这与异种移植物模型有关,其中肿瘤可能很小并且难以与宿主组织分离。我们引入了交叉比对/交叉杂交的新定义,以定性地比较高通量mRNA测序、RNA-Seq和微阵列检测混合“伪异种移植”样本中肿瘤和基质表达的能力,这与-à-vis交叉比对(RNA-Seq)和交叉杂交(微阵列)中的基因和途径有关。样本包括正常小鼠肺癌细胞和人类乳腺癌细胞;这些以固定比例组合,形成25%步的滴定系列。我们的定义确定了特定物种(人类或小鼠)中在同一物种RNA中不可检测表达但在跨物种RNA中可检测表达的基因。我们展示了这种方法的比较价值,并讨论了它在癌症研究中的潜在贡献。结果:我们的方法可以从任何一个物种中鉴定出具有交叉杂交和/或交叉比对特性的基因。令人惊讶的是,使用更简单和更常见的方法(使用“纯”跨物种样本并将所有检测到的基因称为“交叉”)识别出的基因集并不是使用我们的技术识别出的基因的超集。观察到的交叉杂交水平相对较低:在小鼠中检测到的人类基因为5.3%,在人类中检测到的小鼠基因为3.5%。观察到的交叉比对水平实际上与交叉杂交水平相当:在小鼠中检测到6.5%的人类基因,在人类中检测到2.3%的小鼠基因。我们还观察到同源基因的比例相对较高:40.3%的交叉杂交基因和32.2%的交叉比对基因。将基因目录归一化,使用共识编码序列(CCDS) id (Genome Res 19:1316-1323, 2009),我们的结果表明,观察到的交叉杂交水平很低:2.7%的人类CCDS id在小鼠中被检测到,2.4%的小鼠CCDS id在人类中被检测到。使用RNA-Seq数据的交叉比对水平在小鼠中具有可比性,在人类中检测到的小鼠CCDS id为2.2%,在小鼠中检测到的人类CCDS id为9.9%。然而,交叉排列/交叉杂交基因列表中包含许多肿瘤学研究人员特别感兴趣的基因。结论:我们提出的保守定义识别了小鼠中表达可归因于人类RNA的基因,反之亦然,以及揭示了使用更简单但更成熟的方法无法识别的具有交叉比对/交叉杂交特性的基因。受交叉杂交/交叉比对影响的基因的总体百分比很小,但包括肿瘤研究人员感兴趣的基因。使用哪种平台处理混合异种移植物样品,是微阵列还是RNA-Seq,似乎主要是一个成本问题,以及检测和测量感兴趣的特定基因的表达是否可能受到交叉杂交或交叉比对的影响。
Characteristics of cross-hybridization and cross-alignment of expression in pseudo-xenograft samples by RNA-Seq and microarrays.
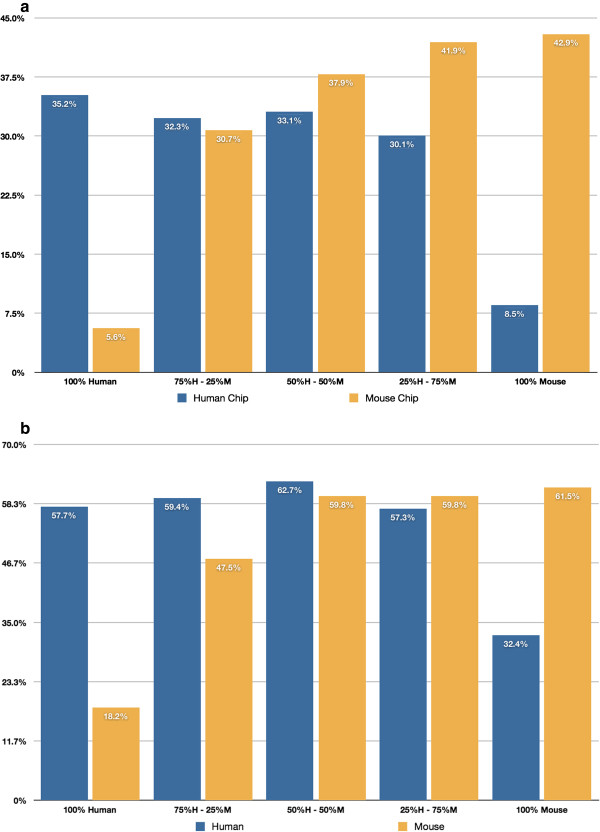
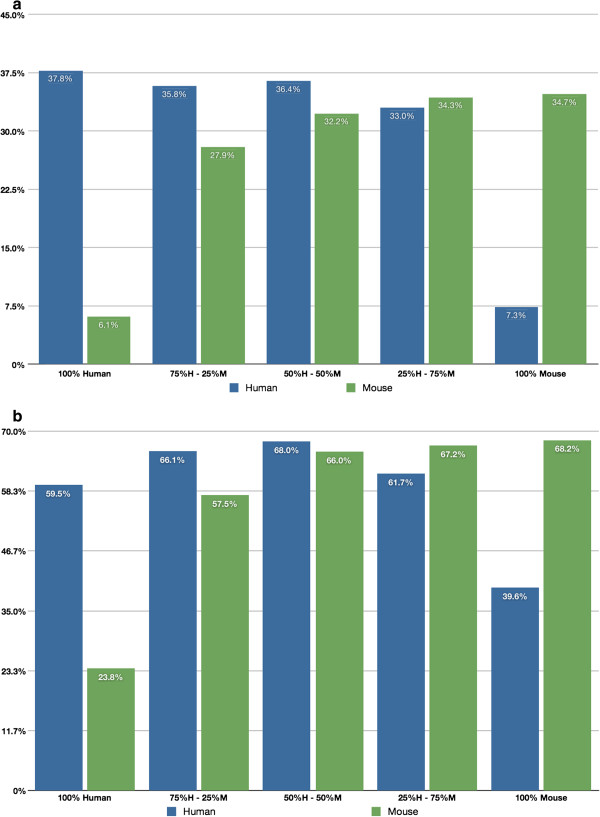
Background: Exploring stromal changes associated with tumor growth and development is a growing area of oncologic research. In order to study molecular changes in the stroma it is recommended to separate tumor tissue from stromal tissue. This is relevant to xenograft models where tumors can be small and difficult to separate from host tissue. We introduce a novel definition of cross-alignment/cross-hybridization to compare qualitatively the ability of high-throughput mRNA sequencing, RNA-Seq, and microarrays to detect tumor and stromal expression from mixed 'pseudo-xenograft' samples vis-à-vis genes and pathways in cross-alignment (RNA-Seq) and cross-hybridization (microarrays). Samples consisted of normal mouse lung and human breast cancer cells; these were combined in fixed proportions to create a titration series of 25% steps. Our definition identifies genes in a given species (human or mouse) with undetectable expression in same-species RNA but detectable expression in cross-species RNA. We demonstrate the comparative value of this method and discuss its potential contribution in cancer research.
Results: Our method can identify genes from either species that demonstrate cross-hybridization and/or cross-alignment properties. Surprisingly, the set of genes identified using a simpler and more common approach (using a 'pure' cross-species sample and calling all detected genes as 'crossers') is not a superset of the genes identified using our technique. The observed levels of cross-hybridization are relatively low: 5.3% of human genes detected in mouse, and 3.5% of mouse genes detected in human. Observed levels of cross-alignment are practically comparable to the levels of cross-hybridization: 6.5% of human genes detected in mouse, and 2.3% of mouse genes detected in human. We also observed a relatively high percentage of orthologs: 40.3% of cross-hybridizing genes, and 32.2% of cross-aligning genes.Normalizing the gene catalog to use Consensus Coding Sequence (CCDS) IDs (Genome Res 19:1316-1323, 2009), our results show that the observed levels of cross-hybridization are low: 2.7% of human CCDS IDs are detected in mouse, and 2.4% of mouse CCDS IDs are detected in human. Levels of cross-alignment using the RNA-Seq data are comparable for the mouse, 2.2% of mouse CCDS IDs detected in human, and 9.9% of human CCDS IDs detected in mouse. However, the lists of cross-aligning/cross-hybridizing genes contain many that are of specific interest to oncologic researchers.
Conclusions: The conservative definition that we propose identifies genes in mouse whose expression can be attributed to human RNA, and vice versa, as well as revealing genes with cross-alignment/cross-hybridization properties which could not be identified using a simpler but more established approach. The overall percentage of genes affected by cross-hybridization/cross-alignment is small, but includes genes that are of interest to oncologic researchers. Which platform to use with mixed xenograft samples, microarrays or RNA-Seq, appears to be primarily a question of cost and whether the detection and measurement of expression of specific genes of interest are likely to be affected by cross-hybridization or cross-alignment.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
