Jason M Held, Birgit Schilling, Alexandria K D'Souza, Tara Srinivasan, Jessica B Behring, Dylan J Sorensen, Christopher C Benz, Bradford W Gibson
{"title":"基于数据依赖性(MS1)和数据非依赖性(MS2)获取的多重蛋白酶消化的ErbB2肿瘤受体的无标记定量和定位","authors":"Jason M Held, Birgit Schilling, Alexandria K D'Souza, Tara Srinivasan, Jessica B Behring, Dylan J Sorensen, Christopher C Benz, Bradford W Gibson","doi":"10.1155/2013/791985","DOIUrl":null,"url":null,"abstract":"<p><p>The receptor tyrosine kinase ErbB2 is a breast cancer biomarker whose posttranslational modifications (PTMs) are a key indicator of its activation. Quantifying the expression and PTMs of biomarkers such as ErbB2 by selected reaction monitoring (SRM) mass spectrometry has several limitations, including minimal coverage and extensive assay development time. Therefore, we assessed the utility of two high resolution, full scan mass spectrometry approaches, MS1 Filtering and SWATH MS2, for targeted ErbB2 proteomics. Endogenous ErbB2 immunoprecipitated from SK-BR-3 cells was in-gel digested with trypsin, chymotrypsin, Asp-N, or trypsin plus Asp-N in triplicate. Data-dependent acquisition with an AB SCIEX TripleTOF 5600 and MS1 Filtering data processing was used to assess peptide and PTM coverage as well as the reproducibility of enzyme digestion. Data-independent acquisition (SWATH) was also performed for MS2 quantitation. MS1 Filtering and SWATH MS2 allow quantitation of all detected analytes after acquisition, enabling the use of multiple proteases for quantitative assessment of target proteins. Combining high resolution proteomics with multiprotease digestion enabled quantitative mapping of ErbB2 with excellent reproducibility, improved amino acid sequence and PTM coverage, and decreased assay development time compared to typical SRM assays. These results demonstrate that high resolution quantitative proteomic approaches are an effective tool for targeted biomarker quantitation.</p>","PeriodicalId":73474,"journal":{"name":"International journal of proteomics","volume":"2013 ","pages":"791985"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/791985","citationCount":"25","resultStr":"{\"title\":\"Label-Free Quantitation and Mapping of the ErbB2 Tumor Receptor by Multiple Protease Digestion with Data-Dependent (MS1) and Data-Independent (MS2) Acquisitions.\",\"authors\":\"Jason M Held, Birgit Schilling, Alexandria K D'Souza, Tara Srinivasan, Jessica B Behring, Dylan J Sorensen, Christopher C Benz, Bradford W Gibson\",\"doi\":\"10.1155/2013/791985\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The receptor tyrosine kinase ErbB2 is a breast cancer biomarker whose posttranslational modifications (PTMs) are a key indicator of its activation. Quantifying the expression and PTMs of biomarkers such as ErbB2 by selected reaction monitoring (SRM) mass spectrometry has several limitations, including minimal coverage and extensive assay development time. Therefore, we assessed the utility of two high resolution, full scan mass spectrometry approaches, MS1 Filtering and SWATH MS2, for targeted ErbB2 proteomics. Endogenous ErbB2 immunoprecipitated from SK-BR-3 cells was in-gel digested with trypsin, chymotrypsin, Asp-N, or trypsin plus Asp-N in triplicate. Data-dependent acquisition with an AB SCIEX TripleTOF 5600 and MS1 Filtering data processing was used to assess peptide and PTM coverage as well as the reproducibility of enzyme digestion. Data-independent acquisition (SWATH) was also performed for MS2 quantitation. MS1 Filtering and SWATH MS2 allow quantitation of all detected analytes after acquisition, enabling the use of multiple proteases for quantitative assessment of target proteins. Combining high resolution proteomics with multiprotease digestion enabled quantitative mapping of ErbB2 with excellent reproducibility, improved amino acid sequence and PTM coverage, and decreased assay development time compared to typical SRM assays. These results demonstrate that high resolution quantitative proteomic approaches are an effective tool for targeted biomarker quantitation.</p>\",\"PeriodicalId\":73474,\"journal\":{\"name\":\"International journal of proteomics\",\"volume\":\"2013 \",\"pages\":\"791985\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2013/791985\",\"citationCount\":\"25\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International journal of proteomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/791985\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/4/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/791985","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/4/4 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Label-Free Quantitation and Mapping of the ErbB2 Tumor Receptor by Multiple Protease Digestion with Data-Dependent (MS1) and Data-Independent (MS2) Acquisitions.
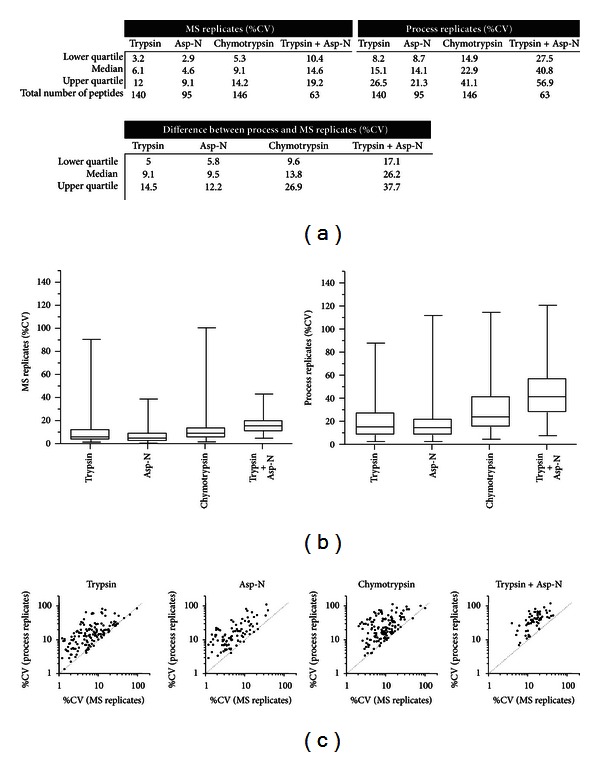
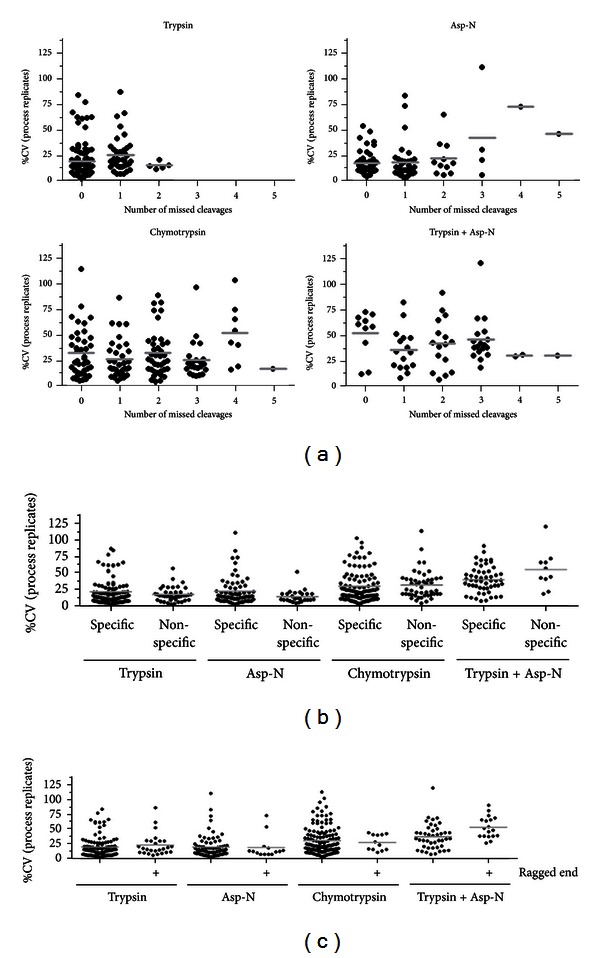
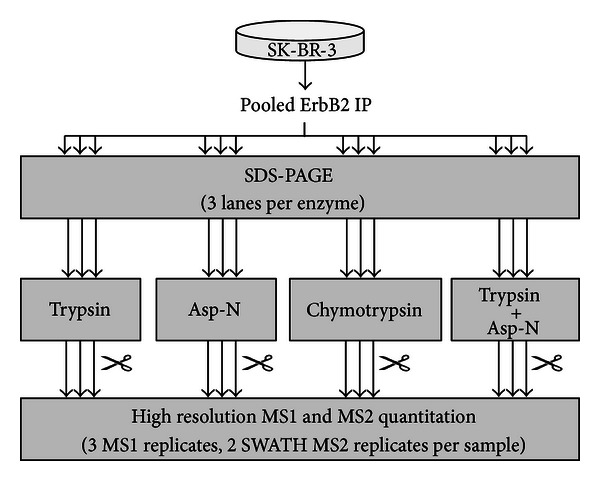
The receptor tyrosine kinase ErbB2 is a breast cancer biomarker whose posttranslational modifications (PTMs) are a key indicator of its activation. Quantifying the expression and PTMs of biomarkers such as ErbB2 by selected reaction monitoring (SRM) mass spectrometry has several limitations, including minimal coverage and extensive assay development time. Therefore, we assessed the utility of two high resolution, full scan mass spectrometry approaches, MS1 Filtering and SWATH MS2, for targeted ErbB2 proteomics. Endogenous ErbB2 immunoprecipitated from SK-BR-3 cells was in-gel digested with trypsin, chymotrypsin, Asp-N, or trypsin plus Asp-N in triplicate. Data-dependent acquisition with an AB SCIEX TripleTOF 5600 and MS1 Filtering data processing was used to assess peptide and PTM coverage as well as the reproducibility of enzyme digestion. Data-independent acquisition (SWATH) was also performed for MS2 quantitation. MS1 Filtering and SWATH MS2 allow quantitation of all detected analytes after acquisition, enabling the use of multiple proteases for quantitative assessment of target proteins. Combining high resolution proteomics with multiprotease digestion enabled quantitative mapping of ErbB2 with excellent reproducibility, improved amino acid sequence and PTM coverage, and decreased assay development time compared to typical SRM assays. These results demonstrate that high resolution quantitative proteomic approaches are an effective tool for targeted biomarker quantitation.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
