Sanvesh Srivastava, Wenyi Wang, Ganiraju Manyam, Carlos Ordonez, Veerabhadran Baladandayuthapani
{"title":"利用层次贝叶斯相关向量机集成多平台基因组数据。","authors":"Sanvesh Srivastava, Wenyi Wang, Ganiraju Manyam, Carlos Ordonez, Veerabhadran Baladandayuthapani","doi":"10.1186/1687-4153-2013-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Recent advances in genome technologies and the subsequent collection of genomic information at various molecular resolutions hold promise to accelerate the discovery of new therapeutic targets. A critical step in achieving these goals is to develop efficient clinical prediction models that integrate these diverse sources of high-throughput data. This step is challenging due to the presence of high-dimensionality and complex interactions in the data. For predicting relevant clinical outcomes, we propose a flexible statistical machine learning approach that acknowledges and models the interaction between platform-specific measurements through nonlinear kernel machines and borrows information within and between platforms through a hierarchical Bayesian framework. Our model has parameters with direct interpretations in terms of the effects of platforms and data interactions within and across platforms. The parameter estimation algorithm in our model uses a computationally efficient variational Bayes approach that scales well to large high-throughput datasets.</p><p><strong>Results: </strong>We apply our methods of integrating gene/mRNA expression and microRNA profiles for predicting patient survival times to The Cancer Genome Atlas (TCGA) based glioblastoma multiforme (GBM) dataset. In terms of prediction accuracy, we show that our non-linear and interaction-based integrative methods perform better than linear alternatives and non-integrative methods that do not account for interactions between the platforms. We also find several prognostic mRNAs and microRNAs that are related to tumor invasion and are known to drive tumor metastasis and severe inflammatory response in GBM. In addition, our analysis reveals several interesting mRNA and microRNA interactions that have known implications in the etiology of GBM.</p><p><strong>Conclusions: </strong>Our approach gains its flexibility and power by modeling the non-linear interaction structures between and within the platforms. Our framework is a useful tool for biomedical researchers, since clinical prediction using multi-platform genomic information is an important step towards personalized treatment of many cancers. We have a freely available software at: http://odin.mdacc.tmc.edu/~vbaladan.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2013 1","pages":"9"},"PeriodicalIF":0.0000,"publicationDate":"2013-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2013-9","citationCount":"9","resultStr":"{\"title\":\"Integrating multi-platform genomic data using hierarchical Bayesian relevance vector machines.\",\"authors\":\"Sanvesh Srivastava, Wenyi Wang, Ganiraju Manyam, Carlos Ordonez, Veerabhadran Baladandayuthapani\",\"doi\":\"10.1186/1687-4153-2013-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Recent advances in genome technologies and the subsequent collection of genomic information at various molecular resolutions hold promise to accelerate the discovery of new therapeutic targets. A critical step in achieving these goals is to develop efficient clinical prediction models that integrate these diverse sources of high-throughput data. This step is challenging due to the presence of high-dimensionality and complex interactions in the data. For predicting relevant clinical outcomes, we propose a flexible statistical machine learning approach that acknowledges and models the interaction between platform-specific measurements through nonlinear kernel machines and borrows information within and between platforms through a hierarchical Bayesian framework. Our model has parameters with direct interpretations in terms of the effects of platforms and data interactions within and across platforms. The parameter estimation algorithm in our model uses a computationally efficient variational Bayes approach that scales well to large high-throughput datasets.</p><p><strong>Results: </strong>We apply our methods of integrating gene/mRNA expression and microRNA profiles for predicting patient survival times to The Cancer Genome Atlas (TCGA) based glioblastoma multiforme (GBM) dataset. In terms of prediction accuracy, we show that our non-linear and interaction-based integrative methods perform better than linear alternatives and non-integrative methods that do not account for interactions between the platforms. We also find several prognostic mRNAs and microRNAs that are related to tumor invasion and are known to drive tumor metastasis and severe inflammatory response in GBM. In addition, our analysis reveals several interesting mRNA and microRNA interactions that have known implications in the etiology of GBM.</p><p><strong>Conclusions: </strong>Our approach gains its flexibility and power by modeling the non-linear interaction structures between and within the platforms. Our framework is a useful tool for biomedical researchers, since clinical prediction using multi-platform genomic information is an important step towards personalized treatment of many cancers. We have a freely available software at: http://odin.mdacc.tmc.edu/~vbaladan.</p>\",\"PeriodicalId\":72957,\"journal\":{\"name\":\"EURASIP journal on bioinformatics & systems biology\",\"volume\":\"2013 1\",\"pages\":\"9\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-06-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1687-4153-2013-9\",\"citationCount\":\"9\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EURASIP journal on bioinformatics & systems biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1687-4153-2013-9\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2013-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Integrating multi-platform genomic data using hierarchical Bayesian relevance vector machines.
Background: Recent advances in genome technologies and the subsequent collection of genomic information at various molecular resolutions hold promise to accelerate the discovery of new therapeutic targets. A critical step in achieving these goals is to develop efficient clinical prediction models that integrate these diverse sources of high-throughput data. This step is challenging due to the presence of high-dimensionality and complex interactions in the data. For predicting relevant clinical outcomes, we propose a flexible statistical machine learning approach that acknowledges and models the interaction between platform-specific measurements through nonlinear kernel machines and borrows information within and between platforms through a hierarchical Bayesian framework. Our model has parameters with direct interpretations in terms of the effects of platforms and data interactions within and across platforms. The parameter estimation algorithm in our model uses a computationally efficient variational Bayes approach that scales well to large high-throughput datasets.
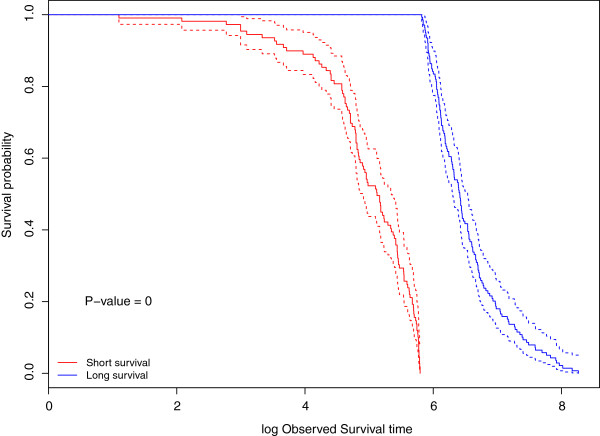
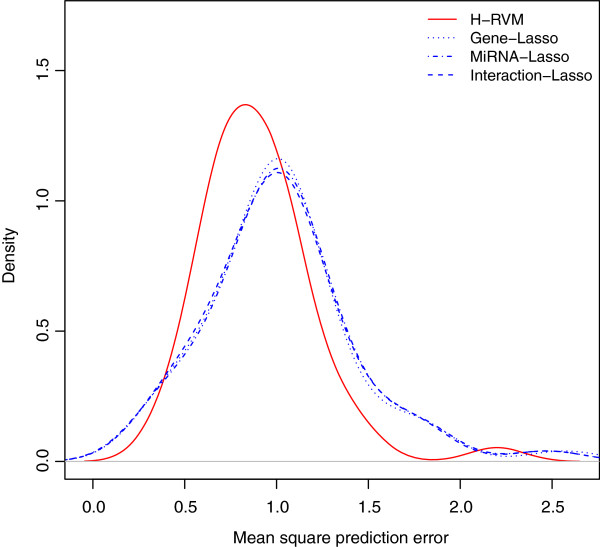
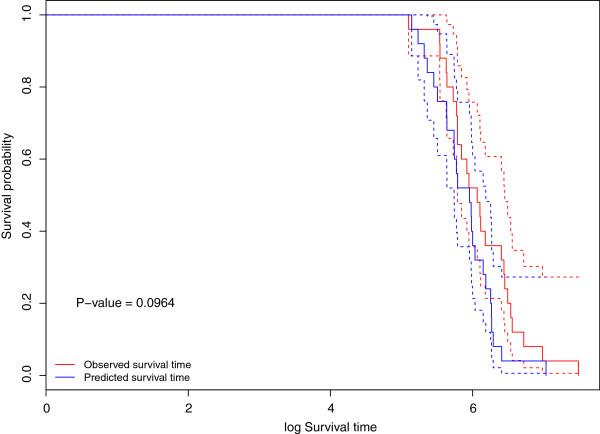
Results: We apply our methods of integrating gene/mRNA expression and microRNA profiles for predicting patient survival times to The Cancer Genome Atlas (TCGA) based glioblastoma multiforme (GBM) dataset. In terms of prediction accuracy, we show that our non-linear and interaction-based integrative methods perform better than linear alternatives and non-integrative methods that do not account for interactions between the platforms. We also find several prognostic mRNAs and microRNAs that are related to tumor invasion and are known to drive tumor metastasis and severe inflammatory response in GBM. In addition, our analysis reveals several interesting mRNA and microRNA interactions that have known implications in the etiology of GBM.
Conclusions: Our approach gains its flexibility and power by modeling the non-linear interaction structures between and within the platforms. Our framework is a useful tool for biomedical researchers, since clinical prediction using multi-platform genomic information is an important step towards personalized treatment of many cancers. We have a freely available software at: http://odin.mdacc.tmc.edu/~vbaladan.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
