{"title":"通过单细胞 mRNA 扩增进行全 RNA 测序的可行性。","authors":"Yunbo Xu, Hongliang Hu, Jie Zheng, Biaoru Li","doi":"10.1155/2013/724124","DOIUrl":null,"url":null,"abstract":"<p><p>Single-cell sampling with RNA-seq analysis plays an important role in reference laboratory; cytogenomic diagnosis for specimens on glass-slides or rare cells in circulating blood for tumor and genetic diseases; measurement of sensitivity and specificity in tumor-tissue genomic analysis with mixed-cells; mechanism analysis of differentiation and proliferation of cancer stem cell for academic purpose. Our single- cell RNA-seq technique shows that fragments were 250-450 bp after fragmentation, amplification, and adapter addition. There were 11.6 million reads mapped in raw sequencing reads (19.6 million). The numbers of mapped genes, mapped transcripts, and mapped exons were 31,332, 41,210, and 85,786, respectively. All QC results demonstrated that RNA-seq techniques could be used for single-cell genomic performance. Analysis of the mapped genes showed that the number of genes mapped by RNA-seq (6767 genes) was much higher than that of differential display (288 libraries) among similar specimens which we have developed and published. The single-cell RNA-seq can detect gene splicing using different subtype TGF-beta analysis. The results from using Q-rtPCR tests demonstrated that sensitivity is 76% and specificity is 55% from single-cell RNA-seq technique with some gene expression missing (2/8 genes). However, it will be feasible to use RNA-seq techniques to contribute to genomic medicine at single-cell level. </p>","PeriodicalId":37545,"journal":{"name":"Genetics Research International","volume":"2013 ","pages":"724124"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3885331/pdf/","citationCount":"0","resultStr":"{\"title\":\"Feasibility of whole RNA sequencing from single-cell mRNA amplification.\",\"authors\":\"Yunbo Xu, Hongliang Hu, Jie Zheng, Biaoru Li\",\"doi\":\"10.1155/2013/724124\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Single-cell sampling with RNA-seq analysis plays an important role in reference laboratory; cytogenomic diagnosis for specimens on glass-slides or rare cells in circulating blood for tumor and genetic diseases; measurement of sensitivity and specificity in tumor-tissue genomic analysis with mixed-cells; mechanism analysis of differentiation and proliferation of cancer stem cell for academic purpose. Our single- cell RNA-seq technique shows that fragments were 250-450 bp after fragmentation, amplification, and adapter addition. There were 11.6 million reads mapped in raw sequencing reads (19.6 million). The numbers of mapped genes, mapped transcripts, and mapped exons were 31,332, 41,210, and 85,786, respectively. All QC results demonstrated that RNA-seq techniques could be used for single-cell genomic performance. Analysis of the mapped genes showed that the number of genes mapped by RNA-seq (6767 genes) was much higher than that of differential display (288 libraries) among similar specimens which we have developed and published. The single-cell RNA-seq can detect gene splicing using different subtype TGF-beta analysis. The results from using Q-rtPCR tests demonstrated that sensitivity is 76% and specificity is 55% from single-cell RNA-seq technique with some gene expression missing (2/8 genes). However, it will be feasible to use RNA-seq techniques to contribute to genomic medicine at single-cell level. </p>\",\"PeriodicalId\":37545,\"journal\":{\"name\":\"Genetics Research International\",\"volume\":\"2013 \",\"pages\":\"724124\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3885331/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics Research International\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/724124\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/12/23 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Research International","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/724124","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/12/23 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Feasibility of whole RNA sequencing from single-cell mRNA amplification.
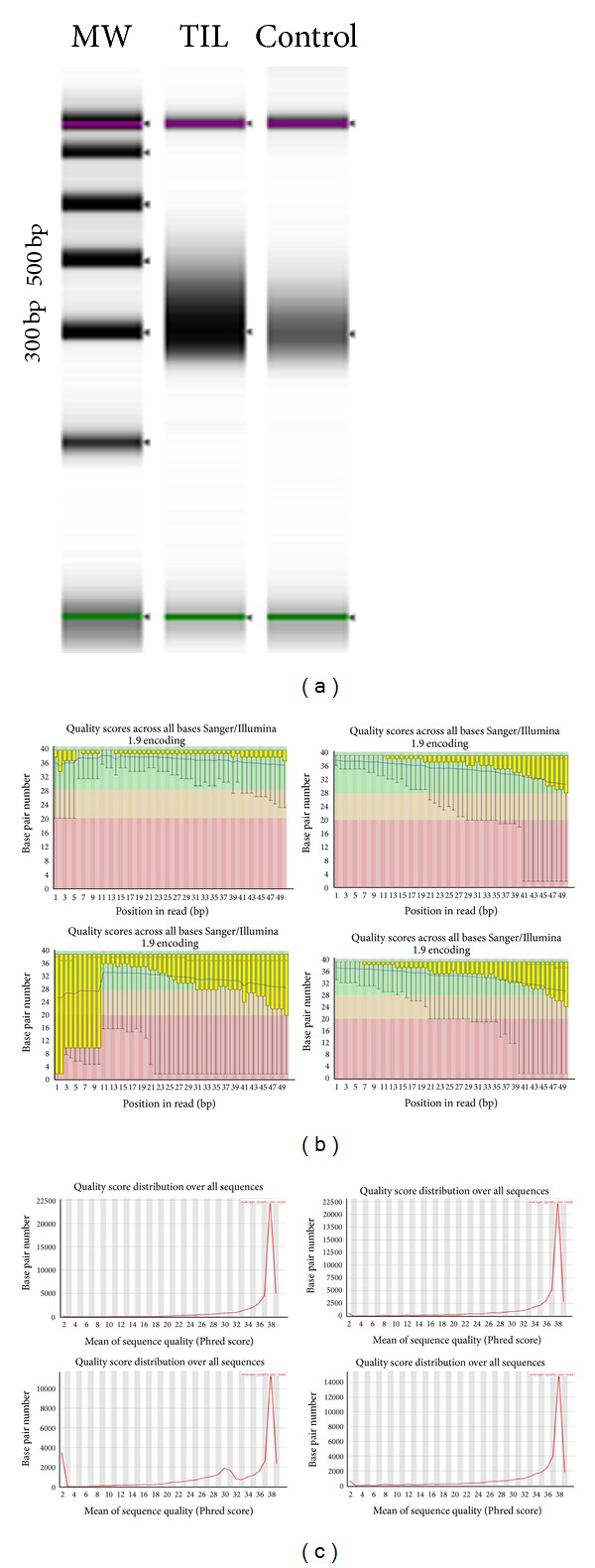
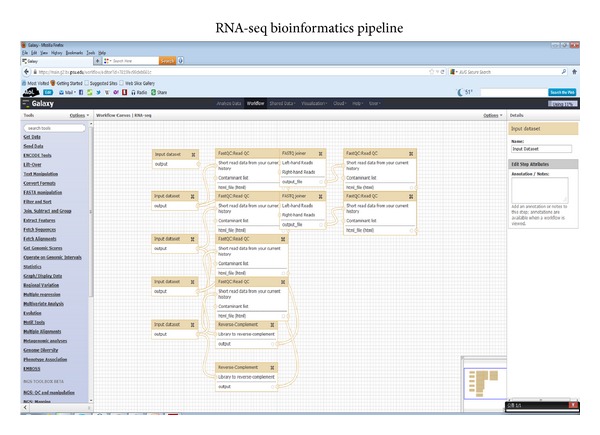
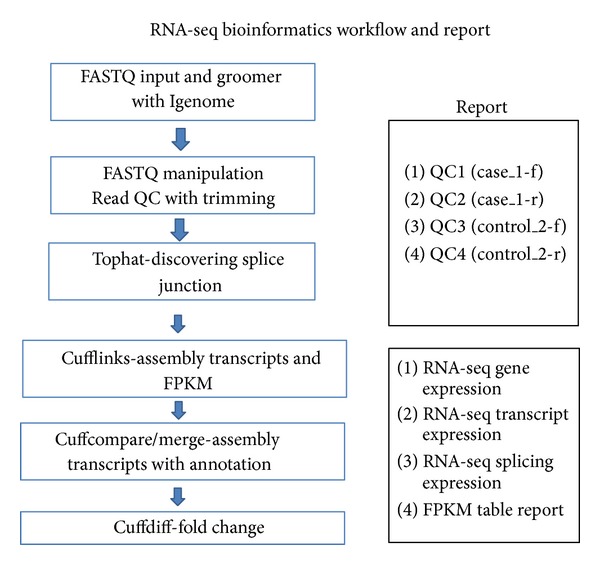
Single-cell sampling with RNA-seq analysis plays an important role in reference laboratory; cytogenomic diagnosis for specimens on glass-slides or rare cells in circulating blood for tumor and genetic diseases; measurement of sensitivity and specificity in tumor-tissue genomic analysis with mixed-cells; mechanism analysis of differentiation and proliferation of cancer stem cell for academic purpose. Our single- cell RNA-seq technique shows that fragments were 250-450 bp after fragmentation, amplification, and adapter addition. There were 11.6 million reads mapped in raw sequencing reads (19.6 million). The numbers of mapped genes, mapped transcripts, and mapped exons were 31,332, 41,210, and 85,786, respectively. All QC results demonstrated that RNA-seq techniques could be used for single-cell genomic performance. Analysis of the mapped genes showed that the number of genes mapped by RNA-seq (6767 genes) was much higher than that of differential display (288 libraries) among similar specimens which we have developed and published. The single-cell RNA-seq can detect gene splicing using different subtype TGF-beta analysis. The results from using Q-rtPCR tests demonstrated that sensitivity is 76% and specificity is 55% from single-cell RNA-seq technique with some gene expression missing (2/8 genes). However, it will be feasible to use RNA-seq techniques to contribute to genomic medicine at single-cell level.
期刊介绍:
Genetics Research International is a peer-reviewed, Open Access journal that publishes original research articles as well as review articles in all areas of genetics and genomics. The journal focuses on articles bearing on heredity, biochemistry, and molecular biology, as well as clinical findings.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
