Frank Krieger, Friedrich Metzger, Sibylle Jablonka
{"title":"对低剂量聚乙二醇化IGF1治疗不敏感的SMARD1小鼠模型的初级运动神经元分化缺陷","authors":"Frank Krieger, Friedrich Metzger, Sibylle Jablonka","doi":"10.4161/rdis.29415","DOIUrl":null,"url":null,"abstract":"<p><p>Muscle atrophy and diaphragmatic palsy are the clinical characteristics of spinal muscular atrophy with respiratory distress type 1 (SMARD1), and are well represented in the neuromuscular degeneration (Nmd(2J) ) mouse, modeling the juvenile form of SMARD1. Both in humans and mice mutations in the IGHMBP2 gene lead to motoneuron degeneration. We could previously demonstrate that treatment with a polyethylene glycol-coupled variant of IGF1 (PEG-IGF1) improves motor functions accompanied by reduced fiber degeneration in the gastrocnemius muscle and the diaphragm, but has no beneficial effect on motoneuron survival. These data raised the question which cell autonomous disease mechanisms contribute to dysfunction and loss of Ighmbp2-deficient motoneurons. An analysis of primary Ighmbp2-deficient motoneurons exhibited differentiation deficits such as reduced spontaneous Ca(2+) transients and altered axon elongation, which was not compensated by PEG-IGF1. This points to an IGF1 independent mechanism of motoneuron degeneration that deserves treatment approaches in addition to IGF1. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"2 ","pages":"e29415"},"PeriodicalIF":0.0000,"publicationDate":"2014-06-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4161/rdis.29415","citationCount":"3","resultStr":"{\"title\":\"Differentiation defects in primary motoneurons from a SMARD1 mouse model that are insensitive to treatment with low dose PEGylated IGF1.\",\"authors\":\"Frank Krieger, Friedrich Metzger, Sibylle Jablonka\",\"doi\":\"10.4161/rdis.29415\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Muscle atrophy and diaphragmatic palsy are the clinical characteristics of spinal muscular atrophy with respiratory distress type 1 (SMARD1), and are well represented in the neuromuscular degeneration (Nmd(2J) ) mouse, modeling the juvenile form of SMARD1. Both in humans and mice mutations in the IGHMBP2 gene lead to motoneuron degeneration. We could previously demonstrate that treatment with a polyethylene glycol-coupled variant of IGF1 (PEG-IGF1) improves motor functions accompanied by reduced fiber degeneration in the gastrocnemius muscle and the diaphragm, but has no beneficial effect on motoneuron survival. These data raised the question which cell autonomous disease mechanisms contribute to dysfunction and loss of Ighmbp2-deficient motoneurons. An analysis of primary Ighmbp2-deficient motoneurons exhibited differentiation deficits such as reduced spontaneous Ca(2+) transients and altered axon elongation, which was not compensated by PEG-IGF1. This points to an IGF1 independent mechanism of motoneuron degeneration that deserves treatment approaches in addition to IGF1. </p>\",\"PeriodicalId\":74639,\"journal\":{\"name\":\"Rare diseases (Austin, Tex.)\",\"volume\":\"2 \",\"pages\":\"e29415\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-06-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.4161/rdis.29415\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Rare diseases (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4161/rdis.29415\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2014/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4161/rdis.29415","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Differentiation defects in primary motoneurons from a SMARD1 mouse model that are insensitive to treatment with low dose PEGylated IGF1.
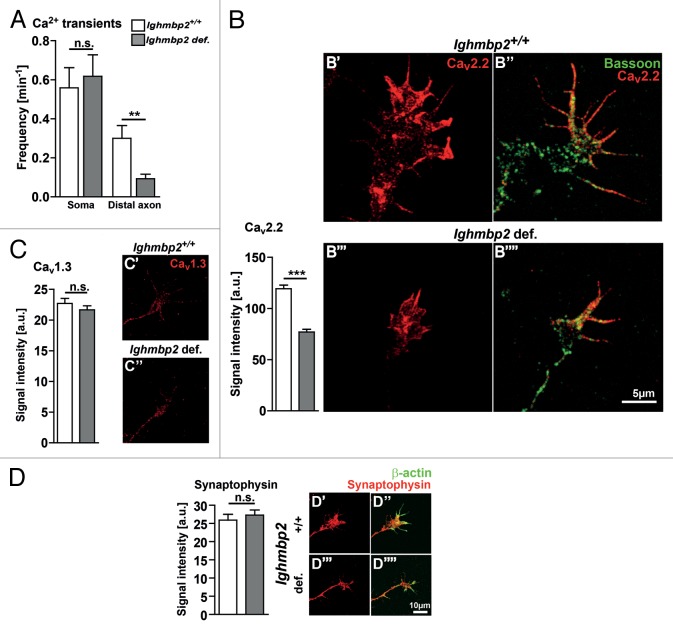
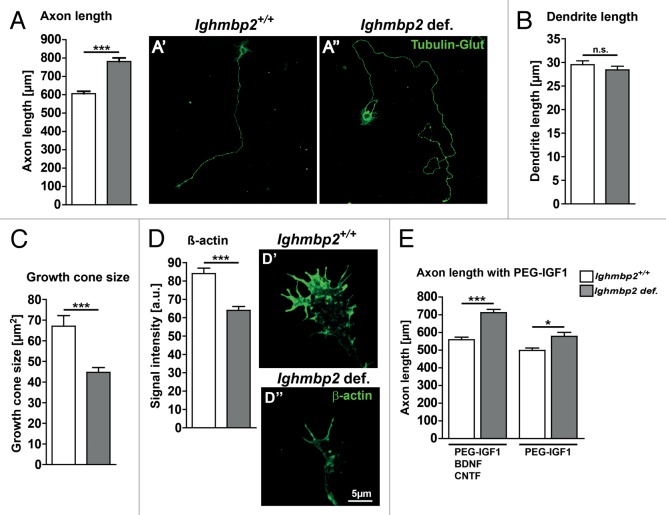
Muscle atrophy and diaphragmatic palsy are the clinical characteristics of spinal muscular atrophy with respiratory distress type 1 (SMARD1), and are well represented in the neuromuscular degeneration (Nmd(2J) ) mouse, modeling the juvenile form of SMARD1. Both in humans and mice mutations in the IGHMBP2 gene lead to motoneuron degeneration. We could previously demonstrate that treatment with a polyethylene glycol-coupled variant of IGF1 (PEG-IGF1) improves motor functions accompanied by reduced fiber degeneration in the gastrocnemius muscle and the diaphragm, but has no beneficial effect on motoneuron survival. These data raised the question which cell autonomous disease mechanisms contribute to dysfunction and loss of Ighmbp2-deficient motoneurons. An analysis of primary Ighmbp2-deficient motoneurons exhibited differentiation deficits such as reduced spontaneous Ca(2+) transients and altered axon elongation, which was not compensated by PEG-IGF1. This points to an IGF1 independent mechanism of motoneuron degeneration that deserves treatment approaches in addition to IGF1.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
