Phanourios Tamamis, Chris A Kieslich, Gregory V Nikiforovich, Trent M Woodruff, Dimitrios Morikis, Georgios Archontis
{"title":"通过隐式溶剂分子动力学模拟和对接研究PMX53抑制C5aR的机制。","authors":"Phanourios Tamamis, Chris A Kieslich, Gregory V Nikiforovich, Trent M Woodruff, Dimitrios Morikis, Georgios Archontis","doi":"10.1186/2046-1682-7-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The complement protein C5a acts by primarily binding and activating the G-protein coupled C5a receptor C5aR (CD88), and is implicated in many inflammatory diseases. The cyclic hexapeptide PMX53 (sequence Ace-Phe-[Orn-Pro-dCha-Trp-Arg]) is a full C5aR antagonist of nanomolar potency, and is widely used to study C5aR function in disease.</p><p><strong>Results: </strong>We construct for the first time molecular models for the C5aR:PMX53 complex without the a priori use of experimental constraints, via a computational framework of molecular dynamics (MD) simulations, docking, conformational clustering and free energy filtering. The models agree with experimental data, and are used to propose important intermolecular interactions contributing to binding, and to develop a hypothesis for the mechanism of PMX53 antagonism.</p><p><strong>Conclusion: </strong>This work forms the basis for the design of improved C5aR antagonists, as well as for atomic-detail mechanistic studies of complement activation and function. Our computational framework can be widely used to develop GPCR-ligand structural models in membrane environments, peptidomimetics and other chemical compounds with potential clinical use.</p>","PeriodicalId":9045,"journal":{"name":"BMC Biophysics","volume":"7 ","pages":"5"},"PeriodicalIF":0.0000,"publicationDate":"2014-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2046-1682-7-5","citationCount":"30","resultStr":"{\"title\":\"Insights into the mechanism of C5aR inhibition by PMX53 via implicit solvent molecular dynamics simulations and docking.\",\"authors\":\"Phanourios Tamamis, Chris A Kieslich, Gregory V Nikiforovich, Trent M Woodruff, Dimitrios Morikis, Georgios Archontis\",\"doi\":\"10.1186/2046-1682-7-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The complement protein C5a acts by primarily binding and activating the G-protein coupled C5a receptor C5aR (CD88), and is implicated in many inflammatory diseases. The cyclic hexapeptide PMX53 (sequence Ace-Phe-[Orn-Pro-dCha-Trp-Arg]) is a full C5aR antagonist of nanomolar potency, and is widely used to study C5aR function in disease.</p><p><strong>Results: </strong>We construct for the first time molecular models for the C5aR:PMX53 complex without the a priori use of experimental constraints, via a computational framework of molecular dynamics (MD) simulations, docking, conformational clustering and free energy filtering. The models agree with experimental data, and are used to propose important intermolecular interactions contributing to binding, and to develop a hypothesis for the mechanism of PMX53 antagonism.</p><p><strong>Conclusion: </strong>This work forms the basis for the design of improved C5aR antagonists, as well as for atomic-detail mechanistic studies of complement activation and function. Our computational framework can be widely used to develop GPCR-ligand structural models in membrane environments, peptidomimetics and other chemical compounds with potential clinical use.</p>\",\"PeriodicalId\":9045,\"journal\":{\"name\":\"BMC Biophysics\",\"volume\":\"7 \",\"pages\":\"5\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/2046-1682-7-5\",\"citationCount\":\"30\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Biophysics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/2046-1682-7-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2014/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biophysics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2046-1682-7-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Insights into the mechanism of C5aR inhibition by PMX53 via implicit solvent molecular dynamics simulations and docking.
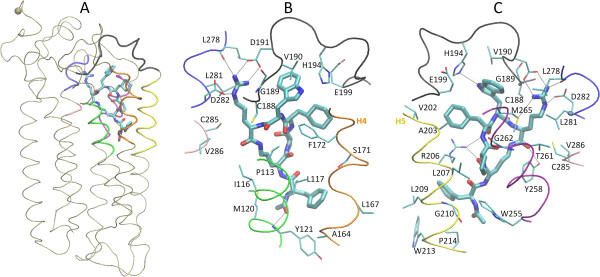
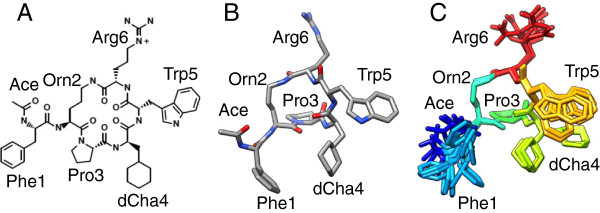
Background: The complement protein C5a acts by primarily binding and activating the G-protein coupled C5a receptor C5aR (CD88), and is implicated in many inflammatory diseases. The cyclic hexapeptide PMX53 (sequence Ace-Phe-[Orn-Pro-dCha-Trp-Arg]) is a full C5aR antagonist of nanomolar potency, and is widely used to study C5aR function in disease.
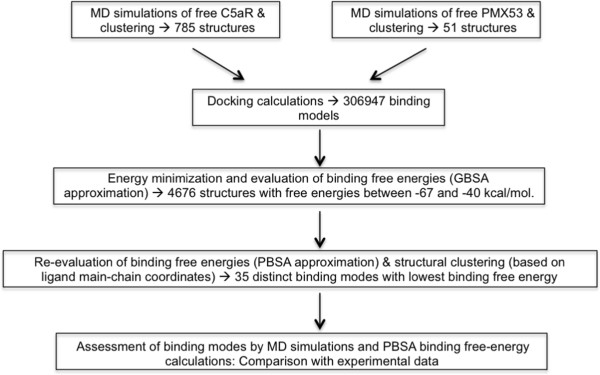
Results: We construct for the first time molecular models for the C5aR:PMX53 complex without the a priori use of experimental constraints, via a computational framework of molecular dynamics (MD) simulations, docking, conformational clustering and free energy filtering. The models agree with experimental data, and are used to propose important intermolecular interactions contributing to binding, and to develop a hypothesis for the mechanism of PMX53 antagonism.
Conclusion: This work forms the basis for the design of improved C5aR antagonists, as well as for atomic-detail mechanistic studies of complement activation and function. Our computational framework can be widely used to develop GPCR-ligand structural models in membrane environments, peptidomimetics and other chemical compounds with potential clinical use.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
