{"title":"VIRAPOPS2支持流感病毒重组。","authors":"Michel Petitjean, Anne Vanet","doi":"10.1186/1751-0473-9-18","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>For over 400 years, due to the reassortment of their segmented genomes, influenza viruses evolve extremely quickly and cause devastating epidemics. This reassortment arises because two flu viruses can infect the same cell and therefore the new virions' genomes will be composed of segment reassortments of the two parental strains. A treatment developed against parents could then be ineffective if the virions' genomes are different enough from their parent's genomes. It is therefore essential to simulate such reassortment phenomena to assess the risk of apparition of new flu strain.</p><p><strong>Findings: </strong>So we decided to upgrade the forward simulator VIRAPOPS, containing already the necessary options to handle non-segmented viral populations. This new version can mimic single or successive reassortments, in birds, humans and/or swines. Other options such as the ability to treat populations of positive or negative sense viral RNAs, were also added. Finally, we propose output options giving statistics of the results.</p><p><strong>Conclusion: </strong>In this paper we present a new version of VIRAPOPS which now manages the viral segment reassortments and the negative sense single strain RNA viruses, these two issues being the cause of serious public health problems.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":"9 ","pages":"18"},"PeriodicalIF":0.0000,"publicationDate":"2014-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1751-0473-9-18","citationCount":"2","resultStr":"{\"title\":\"VIRAPOPS2 supports the influenza virus reassortments.\",\"authors\":\"Michel Petitjean, Anne Vanet\",\"doi\":\"10.1186/1751-0473-9-18\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>For over 400 years, due to the reassortment of their segmented genomes, influenza viruses evolve extremely quickly and cause devastating epidemics. This reassortment arises because two flu viruses can infect the same cell and therefore the new virions' genomes will be composed of segment reassortments of the two parental strains. A treatment developed against parents could then be ineffective if the virions' genomes are different enough from their parent's genomes. It is therefore essential to simulate such reassortment phenomena to assess the risk of apparition of new flu strain.</p><p><strong>Findings: </strong>So we decided to upgrade the forward simulator VIRAPOPS, containing already the necessary options to handle non-segmented viral populations. This new version can mimic single or successive reassortments, in birds, humans and/or swines. Other options such as the ability to treat populations of positive or negative sense viral RNAs, were also added. Finally, we propose output options giving statistics of the results.</p><p><strong>Conclusion: </strong>In this paper we present a new version of VIRAPOPS which now manages the viral segment reassortments and the negative sense single strain RNA viruses, these two issues being the cause of serious public health problems.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\"9 \",\"pages\":\"18\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1751-0473-9-18\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1751-0473-9-18\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2014/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1751-0473-9-18","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
VIRAPOPS2 supports the influenza virus reassortments.
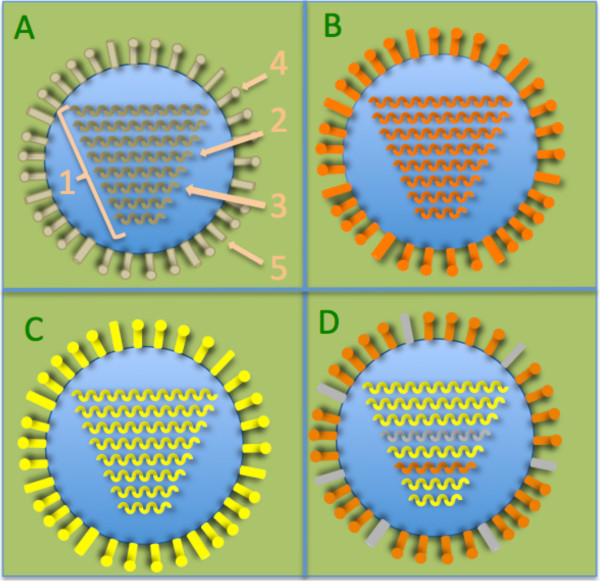
Background: For over 400 years, due to the reassortment of their segmented genomes, influenza viruses evolve extremely quickly and cause devastating epidemics. This reassortment arises because two flu viruses can infect the same cell and therefore the new virions' genomes will be composed of segment reassortments of the two parental strains. A treatment developed against parents could then be ineffective if the virions' genomes are different enough from their parent's genomes. It is therefore essential to simulate such reassortment phenomena to assess the risk of apparition of new flu strain.
Findings: So we decided to upgrade the forward simulator VIRAPOPS, containing already the necessary options to handle non-segmented viral populations. This new version can mimic single or successive reassortments, in birds, humans and/or swines. Other options such as the ability to treat populations of positive or negative sense viral RNAs, were also added. Finally, we propose output options giving statistics of the results.
Conclusion: In this paper we present a new version of VIRAPOPS which now manages the viral segment reassortments and the negative sense single strain RNA viruses, these two issues being the cause of serious public health problems.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
