{"title":"基于AluScan序列的拷贝数变异分析。","authors":"Jian-Feng Yang, Xiao-Fan Ding, Lei Chen, Wai-Kin Mat, Michelle Zhi Xu, Jin-Fei Chen, Jian-Min Wang, Lin Xu, Wai-Sang Poon, Ava Kwong, Gilberto Ka-Kit Leung, Tze-Ching Tan, Chi-Hung Yu, Yue-Bin Ke, Xin-Yun Xu, Xiao-Yan Ke, Ronald Cw Ma, Juliana Cn Chan, Wei-Qing Wan, Li-Wei Zhang, Yogesh Kumar, Shui-Ying Tsang, Shao Li, Hong-Yang Wang, Hong Xue","doi":"10.1186/s13336-014-0015-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>AluScan combines inter-Alu PCR using multiple Alu-based primers with opposite orientations and next-generation sequencing to capture a huge number of Alu-proximal genomic sequences for investigation. Its requirement of only sub-microgram quantities of DNA facilitates the examination of large numbers of samples. However, the special features of AluScan data rendered difficult the calling of copy number variation (CNV) directly using the calling algorithms designed for whole genome sequencing (WGS) or exome sequencing.</p><p><strong>Results: </strong>In this study, an AluScanCNV package has been assembled for efficient CNV calling from AluScan sequencing data employing a Geary-Hinkley transformation (GHT) of read-depth ratios between either paired test-control samples, or between test samples and a reference template constructed from reference samples, to call the localized CNVs, followed by use of a GISTIC-like algorithm to identify recurrent CNVs and circular binary segmentation (CBS) to reveal large extended CNVs. To evaluate the utility of CNVs called from AluScan data, the AluScans from 23 non-cancer and 38 cancer genomes were analyzed in this study. The glioma samples analyzed yielded the familiar extended copy-number losses on chromosomes 1p and 9. Also, the recurrent somatic CNVs identified from liver cancer samples were similar to those reported for liver cancer WGS with respect to a striking enrichment of copy-number gains in chromosomes 1q and 8q. When localized or recurrent CNV-features capable of distinguishing between liver and non-liver cancer samples were selected by correlation-based machine learning, a highly accurate separation of the liver and non-liver cancer classes was attained.</p><p><strong>Conclusions: </strong>The results obtained from non-cancer and cancerous tissues indicated that the AluScanCNV package can be employed to call localized, recurrent and extended CNVs from AluScan sequences. Moreover, both the localized and recurrent CNVs identified by this method could be subjected to machine-learning selection to yield distinguishing CNV-features that were capable of separating between liver cancers and other types of cancers. Since the method is applicable to any human DNA sample with or without the availability of a paired control, it can also be employed to analyze the constitutional CNVs of individuals.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"4 1","pages":"15"},"PeriodicalIF":0.0000,"publicationDate":"2014-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13336-014-0015-z","citationCount":"12","resultStr":"{\"title\":\"Copy number variation analysis based on AluScan sequences.\",\"authors\":\"Jian-Feng Yang, Xiao-Fan Ding, Lei Chen, Wai-Kin Mat, Michelle Zhi Xu, Jin-Fei Chen, Jian-Min Wang, Lin Xu, Wai-Sang Poon, Ava Kwong, Gilberto Ka-Kit Leung, Tze-Ching Tan, Chi-Hung Yu, Yue-Bin Ke, Xin-Yun Xu, Xiao-Yan Ke, Ronald Cw Ma, Juliana Cn Chan, Wei-Qing Wan, Li-Wei Zhang, Yogesh Kumar, Shui-Ying Tsang, Shao Li, Hong-Yang Wang, Hong Xue\",\"doi\":\"10.1186/s13336-014-0015-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>AluScan combines inter-Alu PCR using multiple Alu-based primers with opposite orientations and next-generation sequencing to capture a huge number of Alu-proximal genomic sequences for investigation. Its requirement of only sub-microgram quantities of DNA facilitates the examination of large numbers of samples. However, the special features of AluScan data rendered difficult the calling of copy number variation (CNV) directly using the calling algorithms designed for whole genome sequencing (WGS) or exome sequencing.</p><p><strong>Results: </strong>In this study, an AluScanCNV package has been assembled for efficient CNV calling from AluScan sequencing data employing a Geary-Hinkley transformation (GHT) of read-depth ratios between either paired test-control samples, or between test samples and a reference template constructed from reference samples, to call the localized CNVs, followed by use of a GISTIC-like algorithm to identify recurrent CNVs and circular binary segmentation (CBS) to reveal large extended CNVs. To evaluate the utility of CNVs called from AluScan data, the AluScans from 23 non-cancer and 38 cancer genomes were analyzed in this study. The glioma samples analyzed yielded the familiar extended copy-number losses on chromosomes 1p and 9. Also, the recurrent somatic CNVs identified from liver cancer samples were similar to those reported for liver cancer WGS with respect to a striking enrichment of copy-number gains in chromosomes 1q and 8q. When localized or recurrent CNV-features capable of distinguishing between liver and non-liver cancer samples were selected by correlation-based machine learning, a highly accurate separation of the liver and non-liver cancer classes was attained.</p><p><strong>Conclusions: </strong>The results obtained from non-cancer and cancerous tissues indicated that the AluScanCNV package can be employed to call localized, recurrent and extended CNVs from AluScan sequences. Moreover, both the localized and recurrent CNVs identified by this method could be subjected to machine-learning selection to yield distinguishing CNV-features that were capable of separating between liver cancers and other types of cancers. Since the method is applicable to any human DNA sample with or without the availability of a paired control, it can also be employed to analyze the constitutional CNVs of individuals.</p>\",\"PeriodicalId\":73663,\"journal\":{\"name\":\"Journal of clinical bioinformatics\",\"volume\":\"4 1\",\"pages\":\"15\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-12-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13336-014-0015-z\",\"citationCount\":\"12\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13336-014-0015-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2014/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13336-014-0015-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Copy number variation analysis based on AluScan sequences.
Background: AluScan combines inter-Alu PCR using multiple Alu-based primers with opposite orientations and next-generation sequencing to capture a huge number of Alu-proximal genomic sequences for investigation. Its requirement of only sub-microgram quantities of DNA facilitates the examination of large numbers of samples. However, the special features of AluScan data rendered difficult the calling of copy number variation (CNV) directly using the calling algorithms designed for whole genome sequencing (WGS) or exome sequencing.
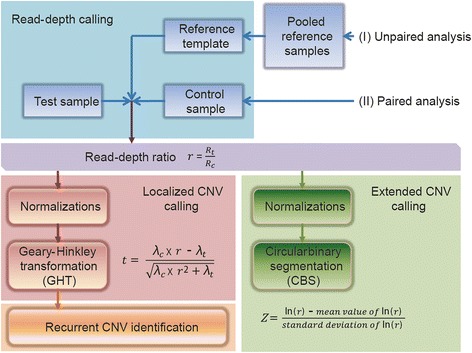
Results: In this study, an AluScanCNV package has been assembled for efficient CNV calling from AluScan sequencing data employing a Geary-Hinkley transformation (GHT) of read-depth ratios between either paired test-control samples, or between test samples and a reference template constructed from reference samples, to call the localized CNVs, followed by use of a GISTIC-like algorithm to identify recurrent CNVs and circular binary segmentation (CBS) to reveal large extended CNVs. To evaluate the utility of CNVs called from AluScan data, the AluScans from 23 non-cancer and 38 cancer genomes were analyzed in this study. The glioma samples analyzed yielded the familiar extended copy-number losses on chromosomes 1p and 9. Also, the recurrent somatic CNVs identified from liver cancer samples were similar to those reported for liver cancer WGS with respect to a striking enrichment of copy-number gains in chromosomes 1q and 8q. When localized or recurrent CNV-features capable of distinguishing between liver and non-liver cancer samples were selected by correlation-based machine learning, a highly accurate separation of the liver and non-liver cancer classes was attained.
Conclusions: The results obtained from non-cancer and cancerous tissues indicated that the AluScanCNV package can be employed to call localized, recurrent and extended CNVs from AluScan sequences. Moreover, both the localized and recurrent CNVs identified by this method could be subjected to machine-learning selection to yield distinguishing CNV-features that were capable of separating between liver cancers and other types of cancers. Since the method is applicable to any human DNA sample with or without the availability of a paired control, it can also be employed to analyze the constitutional CNVs of individuals.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
