{"title":"hmm序列分析的现代计算技术。","authors":"Xiandong Meng, Yanqing Ji","doi":"10.1155/2013/252183","DOIUrl":null,"url":null,"abstract":"<p><p>This paper focuses on the latest research and critical reviews on modern computing architectures, software and hardware accelerated algorithms for bioinformatics data analysis with an emphasis on one of the most important sequence analysis applications-hidden Markov models (HMM). We show the detailed performance comparison of sequence analysis tools on various computing platforms recently developed in the bioinformatics society. The characteristics of the sequence analysis, such as data and compute-intensive natures, make it very attractive to optimize and parallelize by using both traditional software approach and innovated hardware acceleration technologies. </p>","PeriodicalId":90877,"journal":{"name":"ISRN bioinformatics","volume":"2013 ","pages":"252183"},"PeriodicalIF":0.0000,"publicationDate":"2013-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393056/pdf/","citationCount":"21","resultStr":"{\"title\":\"Modern Computational Techniques for the HMMER Sequence Analysis.\",\"authors\":\"Xiandong Meng, Yanqing Ji\",\"doi\":\"10.1155/2013/252183\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>This paper focuses on the latest research and critical reviews on modern computing architectures, software and hardware accelerated algorithms for bioinformatics data analysis with an emphasis on one of the most important sequence analysis applications-hidden Markov models (HMM). We show the detailed performance comparison of sequence analysis tools on various computing platforms recently developed in the bioinformatics society. The characteristics of the sequence analysis, such as data and compute-intensive natures, make it very attractive to optimize and parallelize by using both traditional software approach and innovated hardware acceleration technologies. </p>\",\"PeriodicalId\":90877,\"journal\":{\"name\":\"ISRN bioinformatics\",\"volume\":\"2013 \",\"pages\":\"252183\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4393056/pdf/\",\"citationCount\":\"21\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISRN bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/252183\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/252183","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Modern Computational Techniques for the HMMER Sequence Analysis.
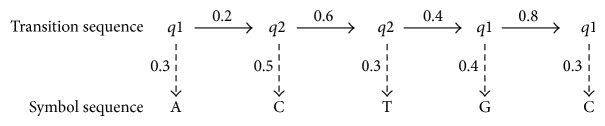
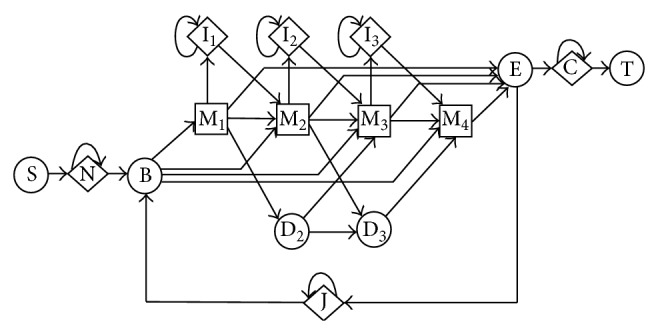
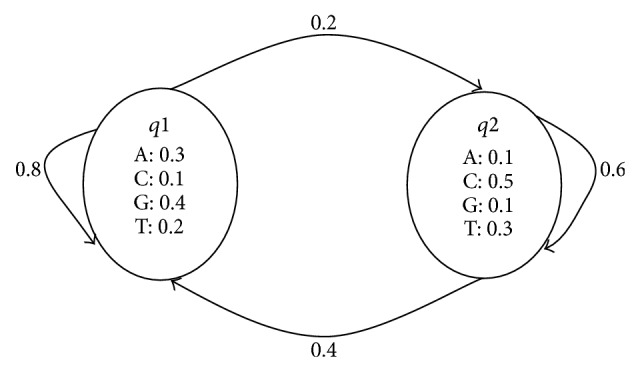
This paper focuses on the latest research and critical reviews on modern computing architectures, software and hardware accelerated algorithms for bioinformatics data analysis with an emphasis on one of the most important sequence analysis applications-hidden Markov models (HMM). We show the detailed performance comparison of sequence analysis tools on various computing platforms recently developed in the bioinformatics society. The characteristics of the sequence analysis, such as data and compute-intensive natures, make it very attractive to optimize and parallelize by using both traditional software approach and innovated hardware acceleration technologies.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
