Claire Mulvey, Bettina Thur, Mark Crawford, Jasminka Godovac-Zimmermann
{"title":"基于Ms/Ms测序方法的定量蛋白质组学缺失了多少蛋白质?","authors":"Claire Mulvey, Bettina Thur, Mark Crawford, Jasminka Godovac-Zimmermann","doi":"10.4137/PRI.S5882","DOIUrl":null,"url":null,"abstract":"<p><p>Current bottom-up quantitative proteomics methods based on MS/MS sequencing of peptides are shown to be strongly dependent on sample preparation. Using cytosolic proteins from MCF-7 breast cancer cells, it is shown that protein pre-fractionation based on pI and MW is more effective than pre-fractionation using only MW in increasing the number of observed proteins (947 vs. 704 proteins) and the number of spectral counts per protein. Combination of MS data from the different pre-fractionation methods results in further improvements (1238 proteins). We discuss that at present the main limitation on quantitation by MS/MS sequencing is not MS sensitivity and protein abundance, but rather extensive peptide overlap and limited MS/MS sequencing throughput, and that this favors internally calibrated methods such as SILAC, ICAT or ITRAQ over spectral counting methods in attempts to drastically improve proteome coverage of biological samples.</p>","PeriodicalId":88975,"journal":{"name":"Proteomics insights","volume":"3 ","pages":"61-66"},"PeriodicalIF":0.0000,"publicationDate":"2010-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4340538/pdf/","citationCount":"0","resultStr":"{\"title\":\"How Many proteins are Missed in Quantitative proteomics Based on Ms/Ms sequencing Methods?\",\"authors\":\"Claire Mulvey, Bettina Thur, Mark Crawford, Jasminka Godovac-Zimmermann\",\"doi\":\"10.4137/PRI.S5882\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Current bottom-up quantitative proteomics methods based on MS/MS sequencing of peptides are shown to be strongly dependent on sample preparation. Using cytosolic proteins from MCF-7 breast cancer cells, it is shown that protein pre-fractionation based on pI and MW is more effective than pre-fractionation using only MW in increasing the number of observed proteins (947 vs. 704 proteins) and the number of spectral counts per protein. Combination of MS data from the different pre-fractionation methods results in further improvements (1238 proteins). We discuss that at present the main limitation on quantitation by MS/MS sequencing is not MS sensitivity and protein abundance, but rather extensive peptide overlap and limited MS/MS sequencing throughput, and that this favors internally calibrated methods such as SILAC, ICAT or ITRAQ over spectral counting methods in attempts to drastically improve proteome coverage of biological samples.</p>\",\"PeriodicalId\":88975,\"journal\":{\"name\":\"Proteomics insights\",\"volume\":\"3 \",\"pages\":\"61-66\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2010-10-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4340538/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteomics insights\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4137/PRI.S5882\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4137/PRI.S5882","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
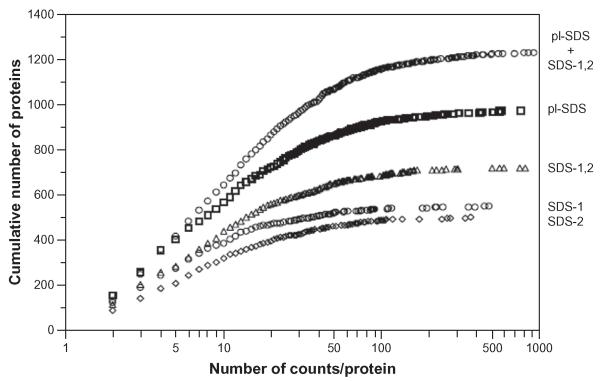
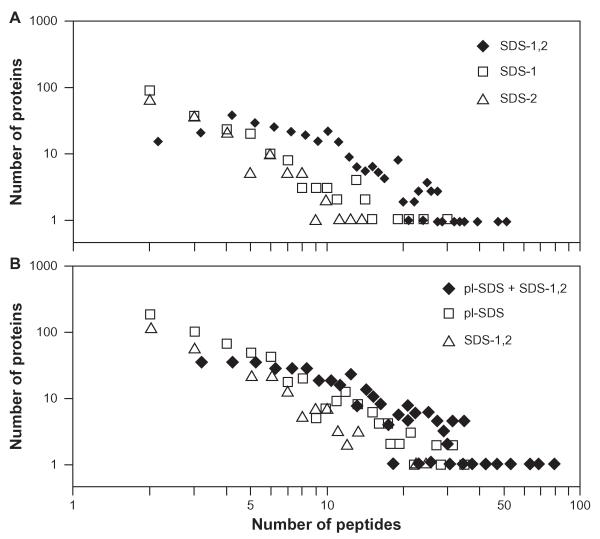
目前基于MS/MS测序的自底向上定量蛋白质组学方法强烈依赖于样品制备。使用MCF-7乳腺癌细胞的细胞质蛋白,研究表明,在增加观察到的蛋白数量(947 vs 704)和每个蛋白的光谱计数数量方面,基于pI和MW的蛋白预分离比仅使用MW的蛋白预分离更有效。结合来自不同预分离方法的质谱数据,进一步改进(1238个蛋白)。我们讨论了目前MS/MS测序定量的主要限制不是MS敏感性和蛋白质丰度,而是广泛的肽重叠和有限的MS/MS测序通量,这有利于内部校准方法,如SILAC, ICAT或ITRAQ,而不是光谱计数方法,试图大幅提高生物样品的蛋白质组覆盖率。
How Many proteins are Missed in Quantitative proteomics Based on Ms/Ms sequencing Methods?
Current bottom-up quantitative proteomics methods based on MS/MS sequencing of peptides are shown to be strongly dependent on sample preparation. Using cytosolic proteins from MCF-7 breast cancer cells, it is shown that protein pre-fractionation based on pI and MW is more effective than pre-fractionation using only MW in increasing the number of observed proteins (947 vs. 704 proteins) and the number of spectral counts per protein. Combination of MS data from the different pre-fractionation methods results in further improvements (1238 proteins). We discuss that at present the main limitation on quantitation by MS/MS sequencing is not MS sensitivity and protein abundance, but rather extensive peptide overlap and limited MS/MS sequencing throughput, and that this favors internally calibrated methods such as SILAC, ICAT or ITRAQ over spectral counting methods in attempts to drastically improve proteome coverage of biological samples.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
