Bryce Kille, Advait Balaji, Fritz J Sedlazeck, Michael Nute, Todd J Treangen
{"title":"端粒与端粒组装时代的多基因组比对。","authors":"Bryce Kille, Advait Balaji, Fritz J Sedlazeck, Michael Nute, Todd J Treangen","doi":"10.1186/s13059-022-02735-6","DOIUrl":null,"url":null,"abstract":"<p><p>With the arrival of telomere-to-telomere (T2T) assemblies of the human genome comes the computational challenge of efficiently and accurately constructing multiple genome alignments at an unprecedented scale. By identifying nucleotides across genomes which share a common ancestor, multiple genome alignments commonly serve as the bedrock for comparative genomics studies. In this review, we provide an overview of the algorithmic template that most multiple genome alignment methods follow. We also discuss prospective areas of improvement of multiple genome alignment for keeping up with continuously arriving high-quality T2T assembled genomes and for unlocking clinically-relevant insights.</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"23 1","pages":"182"},"PeriodicalIF":12.3000,"publicationDate":"2022-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9421119/pdf/","citationCount":"13","resultStr":"{\"title\":\"Multiple genome alignment in the telomere-to-telomere assembly era.\",\"authors\":\"Bryce Kille, Advait Balaji, Fritz J Sedlazeck, Michael Nute, Todd J Treangen\",\"doi\":\"10.1186/s13059-022-02735-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>With the arrival of telomere-to-telomere (T2T) assemblies of the human genome comes the computational challenge of efficiently and accurately constructing multiple genome alignments at an unprecedented scale. By identifying nucleotides across genomes which share a common ancestor, multiple genome alignments commonly serve as the bedrock for comparative genomics studies. In this review, we provide an overview of the algorithmic template that most multiple genome alignment methods follow. We also discuss prospective areas of improvement of multiple genome alignment for keeping up with continuously arriving high-quality T2T assembled genomes and for unlocking clinically-relevant insights.</p>\",\"PeriodicalId\":48922,\"journal\":{\"name\":\"Genome Biology\",\"volume\":\"23 1\",\"pages\":\"182\"},\"PeriodicalIF\":12.3000,\"publicationDate\":\"2022-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9421119/pdf/\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13059-022-02735-6\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-022-02735-6","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Multiple genome alignment in the telomere-to-telomere assembly era.
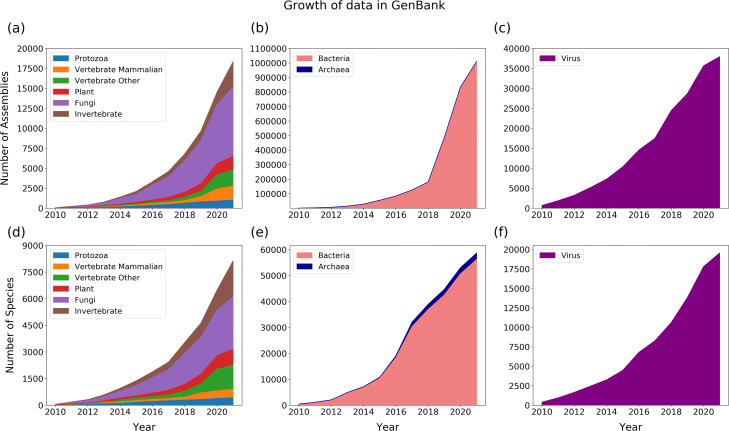
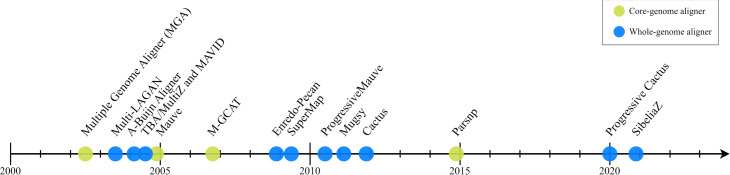
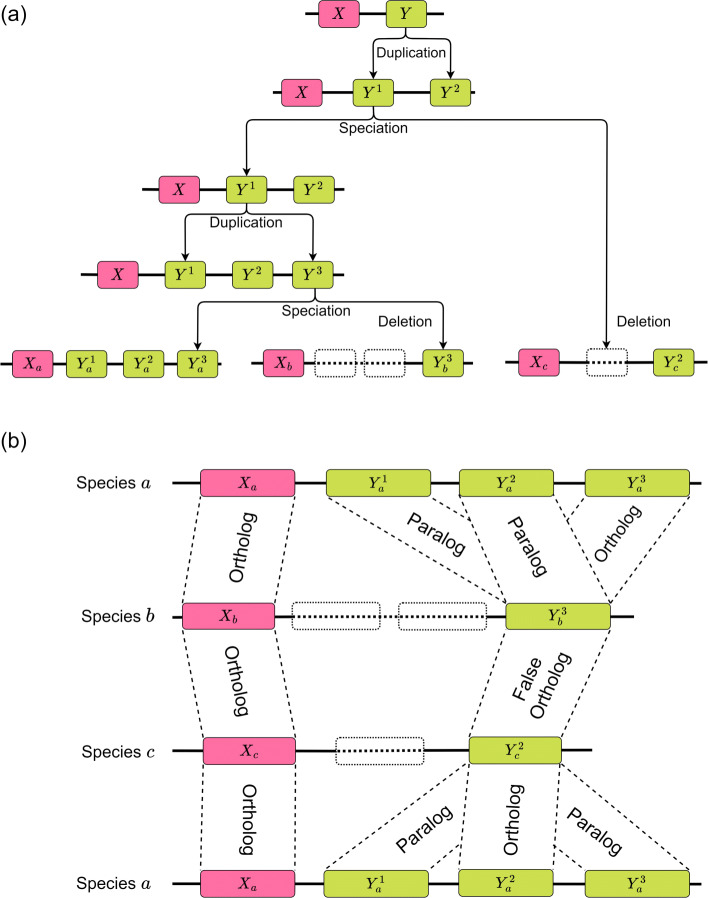
With the arrival of telomere-to-telomere (T2T) assemblies of the human genome comes the computational challenge of efficiently and accurately constructing multiple genome alignments at an unprecedented scale. By identifying nucleotides across genomes which share a common ancestor, multiple genome alignments commonly serve as the bedrock for comparative genomics studies. In this review, we provide an overview of the algorithmic template that most multiple genome alignment methods follow. We also discuss prospective areas of improvement of multiple genome alignment for keeping up with continuously arriving high-quality T2T assembled genomes and for unlocking clinically-relevant insights.
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
