Hana Jeong, Hyeyoung Yoon, Yerin Lee, Jun Tae Kim, Moses Yang, Gayoung Kim, Bom Jung, Seok Hee Park, Choong-Eun Lee
{"title":"SOCS3通过ROS-和p38 mapk依赖通路下调GILZ,减弱地塞米松诱导的M2极化。","authors":"Hana Jeong, Hyeyoung Yoon, Yerin Lee, Jun Tae Kim, Moses Yang, Gayoung Kim, Bom Jung, Seok Hee Park, Choong-Eun Lee","doi":"10.4110/in.2022.22.e33","DOIUrl":null,"url":null,"abstract":"<p><p>Suppressors of cytokine signaling (SOCS) have emerged as potential regulators of macrophage function. We have investigated mechanisms of SOCS3 action on type 2 macrophage (M2) differentiation induced by glucocorticoid using human monocytic cell lines and mouse bone marrow-derived macrophages. Treatment of THP1 monocytic cells with dexamethasone (Dex) induced ROS generation and M2 polarization promoting IL-10 and TGF-β production, while suppressing IL-1β, TNF-α and IL-6 production. SOCS3 over-expression reduced, whereas SOCS3 ablation enhanced IL-10 and TGF-β induction with concomitant regulation of ROS. As a mediator of M2 differentiation, glucocorticoid-induced leucine zipper (GILZ) was down-regulated by SOCS3 and up-regulated by shSOCS3. The induction of GILZ and IL-10 by Dex was dependent on ROS and p38 MAPK activity. Importantly, GILZ ablation led to the inhibition of ROS generation and anti-inflammatory cytokine induction by Dex. Moreover, GILZ knock-down negated the up-regulation of IL-10 production induced by shSOCS3 transduction. Our data suggest that SOCS3 targets ROS- and p38-dependent GILZ expression to suppress Dex-induced M2 polarization.</p>","PeriodicalId":13307,"journal":{"name":"Immune Network","volume":"22 4","pages":"e33"},"PeriodicalIF":4.1000,"publicationDate":"2022-06-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d8/c3/in-22-e33.PMC9433193.pdf","citationCount":"4","resultStr":"{\"title\":\"SOCS3 Attenuates Dexamethasone-Induced M2 Polarization by Down-Regulation of GILZ via ROS- and p38 MAPK-Dependent Pathways.\",\"authors\":\"Hana Jeong, Hyeyoung Yoon, Yerin Lee, Jun Tae Kim, Moses Yang, Gayoung Kim, Bom Jung, Seok Hee Park, Choong-Eun Lee\",\"doi\":\"10.4110/in.2022.22.e33\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Suppressors of cytokine signaling (SOCS) have emerged as potential regulators of macrophage function. We have investigated mechanisms of SOCS3 action on type 2 macrophage (M2) differentiation induced by glucocorticoid using human monocytic cell lines and mouse bone marrow-derived macrophages. Treatment of THP1 monocytic cells with dexamethasone (Dex) induced ROS generation and M2 polarization promoting IL-10 and TGF-β production, while suppressing IL-1β, TNF-α and IL-6 production. SOCS3 over-expression reduced, whereas SOCS3 ablation enhanced IL-10 and TGF-β induction with concomitant regulation of ROS. As a mediator of M2 differentiation, glucocorticoid-induced leucine zipper (GILZ) was down-regulated by SOCS3 and up-regulated by shSOCS3. The induction of GILZ and IL-10 by Dex was dependent on ROS and p38 MAPK activity. Importantly, GILZ ablation led to the inhibition of ROS generation and anti-inflammatory cytokine induction by Dex. Moreover, GILZ knock-down negated the up-regulation of IL-10 production induced by shSOCS3 transduction. Our data suggest that SOCS3 targets ROS- and p38-dependent GILZ expression to suppress Dex-induced M2 polarization.</p>\",\"PeriodicalId\":13307,\"journal\":{\"name\":\"Immune Network\",\"volume\":\"22 4\",\"pages\":\"e33\"},\"PeriodicalIF\":4.1000,\"publicationDate\":\"2022-06-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d8/c3/in-22-e33.PMC9433193.pdf\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immune Network\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.4110/in.2022.22.e33\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/8/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immune Network","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.4110/in.2022.22.e33","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/8/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
SOCS3 Attenuates Dexamethasone-Induced M2 Polarization by Down-Regulation of GILZ via ROS- and p38 MAPK-Dependent Pathways.
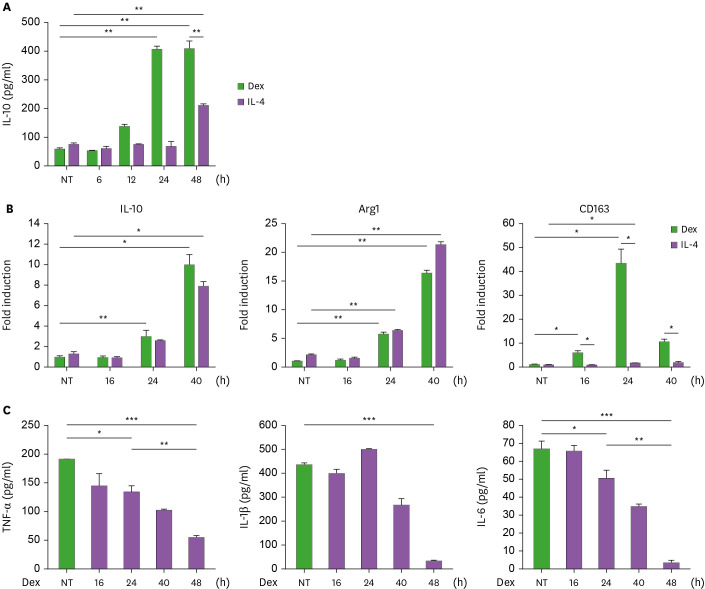
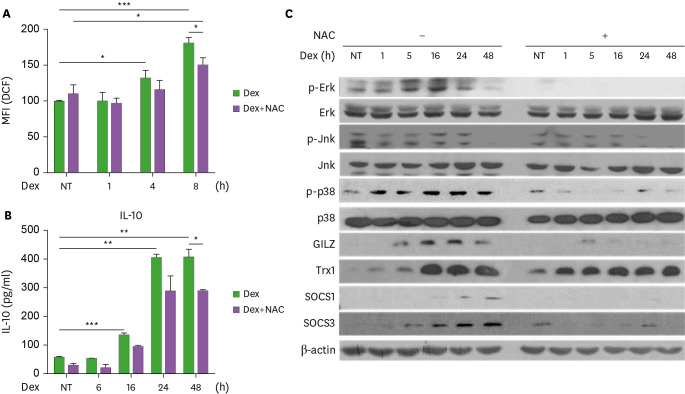
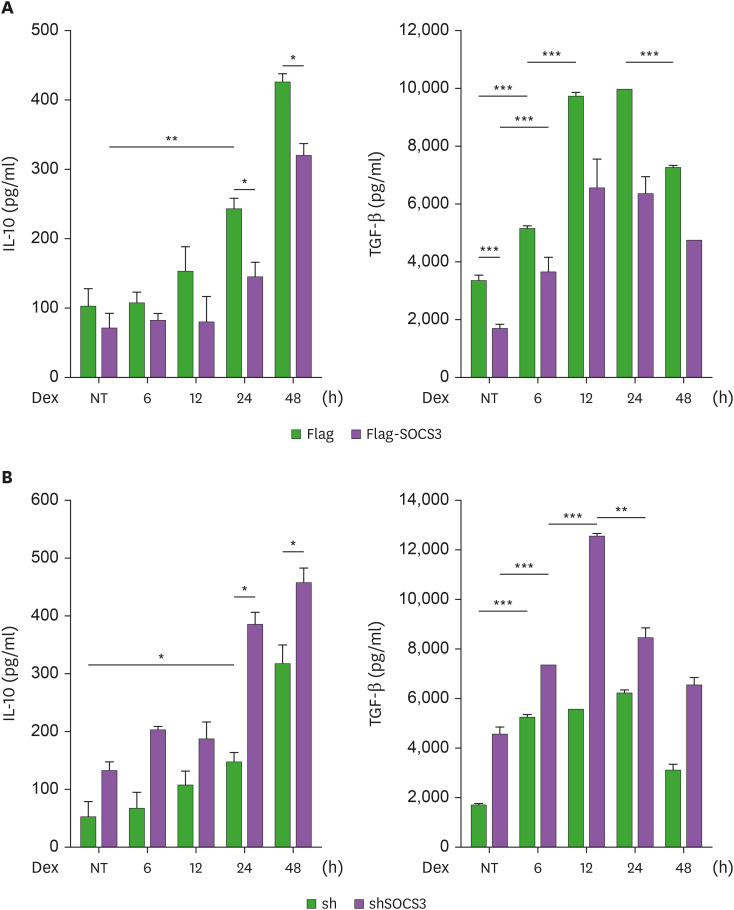
Suppressors of cytokine signaling (SOCS) have emerged as potential regulators of macrophage function. We have investigated mechanisms of SOCS3 action on type 2 macrophage (M2) differentiation induced by glucocorticoid using human monocytic cell lines and mouse bone marrow-derived macrophages. Treatment of THP1 monocytic cells with dexamethasone (Dex) induced ROS generation and M2 polarization promoting IL-10 and TGF-β production, while suppressing IL-1β, TNF-α and IL-6 production. SOCS3 over-expression reduced, whereas SOCS3 ablation enhanced IL-10 and TGF-β induction with concomitant regulation of ROS. As a mediator of M2 differentiation, glucocorticoid-induced leucine zipper (GILZ) was down-regulated by SOCS3 and up-regulated by shSOCS3. The induction of GILZ and IL-10 by Dex was dependent on ROS and p38 MAPK activity. Importantly, GILZ ablation led to the inhibition of ROS generation and anti-inflammatory cytokine induction by Dex. Moreover, GILZ knock-down negated the up-regulation of IL-10 production induced by shSOCS3 transduction. Our data suggest that SOCS3 targets ROS- and p38-dependent GILZ expression to suppress Dex-induced M2 polarization.
期刊介绍:
Immune Network publishes novel findings in basic and clinical immunology and aims to provide a medium through which researchers in various fields of immunology can share and connect. The journal focuses on advances and insights into the regulation of the immune system and the immunological mechanisms of various diseases. Research that provides integrated insights into translational immunology is given preference for publication. All submissions are evaluated based on originality, quality, clarity, and brevity




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
