{"title":"使用内部构建的Java管道挖掘和分析人类冠状病毒基因组中的微卫星。","authors":"P K Bharti, Akhtar Husai","doi":"10.5808/gi.20033","DOIUrl":null,"url":null,"abstract":"<p><p>Microsatellites or simple sequence repeats are motifs of 1 to 6 nucleotides in length present in both coding and non-coding regions of DNA. These are found widely distributed in the whole genome of prokaryotes, eukaryotes, bacteria, and viruses and are used as molecular markers in studying DNA variations, gene regulation, genetic diversity and evolutionary studies, etc. However, in vitro microsatellite identification proves to be time-consuming and expensive. Therefore, the present research has been focused on using an in-house built java pipeline to identify, analyse, design primers and find related statistics of perfect and compound microsatellites in the seven complete genome sequences of coronavirus, including the genome of coronavirus disease 2019, where the host is Homo sapiens. Based on search criteria among seven genomic sequences, it was revealed that the total number of perfect simple sequence repeats (SSRs) found to be in the range of 76 to 118 and compound SSRs from 01 to10, thus reflecting the low conversion of perfect simple sequence to compound repeats. Furthermore, the incidence of SSRs was insignificant but positively correlated with genome size (R2 = 0.45, p > 0.05), with simple sequence repeats relative abundance (R2 = 0.18, p > 0.05) and relative density (R2 = 0.23, p > 0.05). Dinucleotide repeats were the most abundant in the coding region of the genome, followed by tri, mono, and tetra. This comparative study would help us understand the evolutionary relationship, genetic diversity, and hypervariability in minimal time and cost.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":" ","pages":"e35"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9576472/pdf/","citationCount":"0","resultStr":"{\"title\":\"Mining and analysis of microsatellites in human coronavirus genomes using the in-house built Java pipeline.\",\"authors\":\"P K Bharti, Akhtar Husai\",\"doi\":\"10.5808/gi.20033\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Microsatellites or simple sequence repeats are motifs of 1 to 6 nucleotides in length present in both coding and non-coding regions of DNA. These are found widely distributed in the whole genome of prokaryotes, eukaryotes, bacteria, and viruses and are used as molecular markers in studying DNA variations, gene regulation, genetic diversity and evolutionary studies, etc. However, in vitro microsatellite identification proves to be time-consuming and expensive. Therefore, the present research has been focused on using an in-house built java pipeline to identify, analyse, design primers and find related statistics of perfect and compound microsatellites in the seven complete genome sequences of coronavirus, including the genome of coronavirus disease 2019, where the host is Homo sapiens. Based on search criteria among seven genomic sequences, it was revealed that the total number of perfect simple sequence repeats (SSRs) found to be in the range of 76 to 118 and compound SSRs from 01 to10, thus reflecting the low conversion of perfect simple sequence to compound repeats. Furthermore, the incidence of SSRs was insignificant but positively correlated with genome size (R2 = 0.45, p > 0.05), with simple sequence repeats relative abundance (R2 = 0.18, p > 0.05) and relative density (R2 = 0.23, p > 0.05). Dinucleotide repeats were the most abundant in the coding region of the genome, followed by tri, mono, and tetra. This comparative study would help us understand the evolutionary relationship, genetic diversity, and hypervariability in minimal time and cost.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\" \",\"pages\":\"e35\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9576472/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.20033\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/9/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.20033","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
摘要
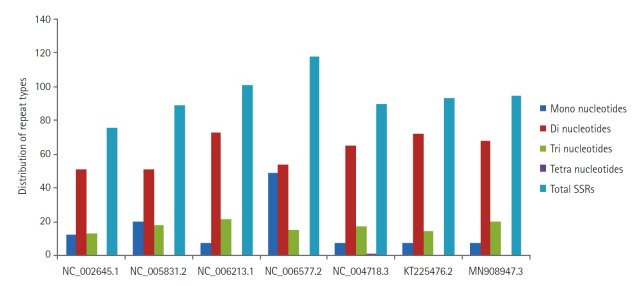
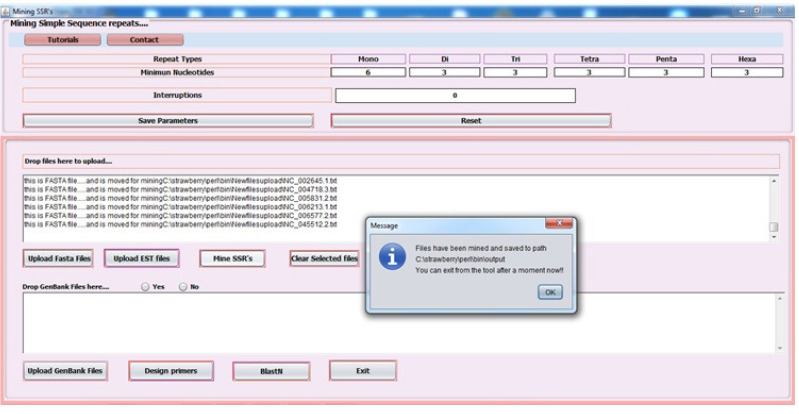
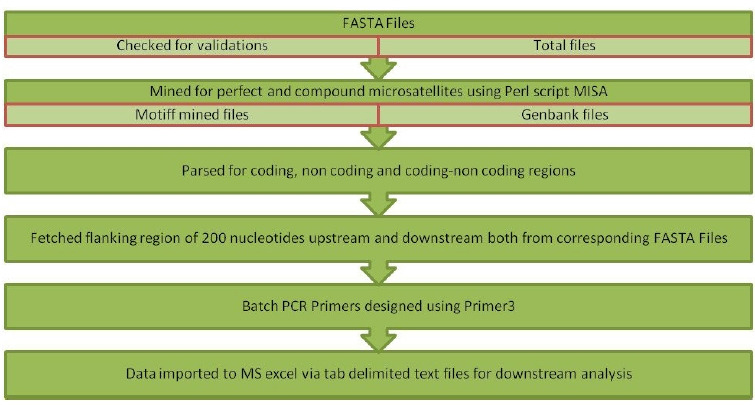
微卫星或简单序列重复是存在于DNA编码区和非编码区长度为1至6个核苷酸的基序。它们广泛存在于原核生物、真核生物、细菌和病毒的全基因组中,被用作研究DNA变异、基因调控、遗传多样性和进化研究等方面的分子标记。然而,体外微卫星鉴定被证明是耗时和昂贵的。因此,本研究的重点是利用内部构建的java流水线,对包括以智人为宿主的2019冠状病毒病基因组在内的7个冠状病毒全基因组序列中的完美微卫星和复合微卫星进行鉴定、分析、设计引物,并进行相关统计。根据7个基因组序列的搜索条件,发现完美简单重复序列(perfect simple sequence repeats, SSRs)的总数在76 ~ 118之间,复合重复序列(compound SSRs)的总数在01 ~ 10之间,反映了完美简单序列到复合重复序列的转化率较低。SSRs的发生率与基因组大小呈正相关(R2 = 0.45, p > 0.05),与简单重复序列的相对丰度(R2 = 0.18, p > 0.05)和相对密度(R2 = 0.23, p > 0.05)。二核苷酸重复序列在基因组编码区最为丰富,其次是三核苷酸重复序列、单核苷酸重复序列和四核苷酸重复序列。这项比较研究将帮助我们在最短的时间和成本内理解进化关系、遗传多样性和高变异性。
Mining and analysis of microsatellites in human coronavirus genomes using the in-house built Java pipeline.
Microsatellites or simple sequence repeats are motifs of 1 to 6 nucleotides in length present in both coding and non-coding regions of DNA. These are found widely distributed in the whole genome of prokaryotes, eukaryotes, bacteria, and viruses and are used as molecular markers in studying DNA variations, gene regulation, genetic diversity and evolutionary studies, etc. However, in vitro microsatellite identification proves to be time-consuming and expensive. Therefore, the present research has been focused on using an in-house built java pipeline to identify, analyse, design primers and find related statistics of perfect and compound microsatellites in the seven complete genome sequences of coronavirus, including the genome of coronavirus disease 2019, where the host is Homo sapiens. Based on search criteria among seven genomic sequences, it was revealed that the total number of perfect simple sequence repeats (SSRs) found to be in the range of 76 to 118 and compound SSRs from 01 to10, thus reflecting the low conversion of perfect simple sequence to compound repeats. Furthermore, the incidence of SSRs was insignificant but positively correlated with genome size (R2 = 0.45, p > 0.05), with simple sequence repeats relative abundance (R2 = 0.18, p > 0.05) and relative density (R2 = 0.23, p > 0.05). Dinucleotide repeats were the most abundant in the coding region of the genome, followed by tri, mono, and tetra. This comparative study would help us understand the evolutionary relationship, genetic diversity, and hypervariability in minimal time and cost.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
