{"title":"cljam:一个用并行处理处理DNA序列比对/图谱(SAM)的库。","authors":"Toshiki Takeuchi, Atsuo Yamada, Takashi Aoki, Kunihiro Nishimura","doi":"10.1186/s13029-016-0058-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Next-generation sequencing can determine DNA bases and the results of sequence alignments are generally stored in files in the Sequence Alignment/Map (SAM) format and the compressed binary version (BAM) of it. SAMtools is a typical tool for dealing with files in the SAM/BAM format. SAMtools has various functions, including detection of variants, visualization of alignments, indexing, extraction of parts of the data and loci, and conversion of file formats. It is written in C and can execute fast. However, SAMtools requires an additional implementation to be used in parallel with, for example, OpenMP (Open Multi-Processing) libraries. For the accumulation of next-generation sequencing data, a simple parallelization program, which can support cloud and PC cluster environments, is required.</p><p><strong>Results: </strong>We have developed cljam using the Clojure programming language, which simplifies parallel programming, to handle SAM/BAM data. Cljam can run in a Java runtime environment (e.g., Windows, Linux, Mac OS X) with Clojure.</p><p><strong>Conclusions: </strong>Cljam can process and analyze SAM/BAM files in parallel and at high speed. The execution time with cljam is almost the same as with SAMtools. The cljam code is written in Clojure and has fewer lines than other similar tools.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":" ","pages":"12"},"PeriodicalIF":0.0000,"publicationDate":"2016-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-016-0058-6","citationCount":"7","resultStr":"{\"title\":\"cljam: a library for handling DNA sequence alignment/map (SAM) with parallel processing.\",\"authors\":\"Toshiki Takeuchi, Atsuo Yamada, Takashi Aoki, Kunihiro Nishimura\",\"doi\":\"10.1186/s13029-016-0058-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Next-generation sequencing can determine DNA bases and the results of sequence alignments are generally stored in files in the Sequence Alignment/Map (SAM) format and the compressed binary version (BAM) of it. SAMtools is a typical tool for dealing with files in the SAM/BAM format. SAMtools has various functions, including detection of variants, visualization of alignments, indexing, extraction of parts of the data and loci, and conversion of file formats. It is written in C and can execute fast. However, SAMtools requires an additional implementation to be used in parallel with, for example, OpenMP (Open Multi-Processing) libraries. For the accumulation of next-generation sequencing data, a simple parallelization program, which can support cloud and PC cluster environments, is required.</p><p><strong>Results: </strong>We have developed cljam using the Clojure programming language, which simplifies parallel programming, to handle SAM/BAM data. Cljam can run in a Java runtime environment (e.g., Windows, Linux, Mac OS X) with Clojure.</p><p><strong>Conclusions: </strong>Cljam can process and analyze SAM/BAM files in parallel and at high speed. The execution time with cljam is almost the same as with SAMtools. The cljam code is written in Clojure and has fewer lines than other similar tools.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\" \",\"pages\":\"12\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-016-0058-6\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-016-0058-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-016-0058-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
引用次数: 7
摘要
背景:下一代测序可以确定DNA碱基,序列比对结果一般存储在序列比对/图谱(sequence Alignment/Map, SAM)格式和压缩二进制版本(BAM)的文件中。SAMtools是处理SAM/BAM格式文件的典型工具。SAMtools具有多种功能,包括检测变体、排列可视化、索引、提取部分数据和轨迹以及转换文件格式。它是用C语言编写的,执行速度很快。然而,SAMtools需要一个额外的实现与OpenMP(开放多处理)库并行使用。为了积累下一代测序数据,需要一个简单的并行化程序,它可以支持云和PC集群环境。结果:我们使用Clojure编程语言开发了cljam来处理SAM/BAM数据,该语言简化了并行编程。Cljam可以通过Clojure在Java运行环境(如Windows、Linux、Mac OS X)中运行。结论:Cljam可以并行、高速地处理和分析SAM/BAM文件。cljam的执行时间与SAMtools几乎相同。cljam代码是用Clojure编写的,比其他类似工具的行数更少。
cljam: a library for handling DNA sequence alignment/map (SAM) with parallel processing.
Background: Next-generation sequencing can determine DNA bases and the results of sequence alignments are generally stored in files in the Sequence Alignment/Map (SAM) format and the compressed binary version (BAM) of it. SAMtools is a typical tool for dealing with files in the SAM/BAM format. SAMtools has various functions, including detection of variants, visualization of alignments, indexing, extraction of parts of the data and loci, and conversion of file formats. It is written in C and can execute fast. However, SAMtools requires an additional implementation to be used in parallel with, for example, OpenMP (Open Multi-Processing) libraries. For the accumulation of next-generation sequencing data, a simple parallelization program, which can support cloud and PC cluster environments, is required.
Results: We have developed cljam using the Clojure programming language, which simplifies parallel programming, to handle SAM/BAM data. Cljam can run in a Java runtime environment (e.g., Windows, Linux, Mac OS X) with Clojure.
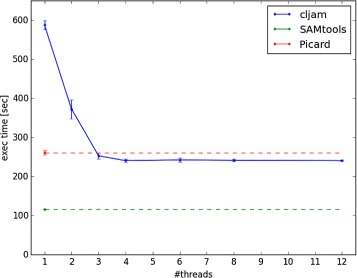
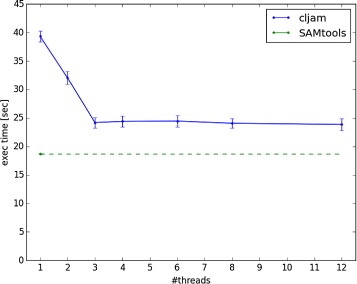
Conclusions: Cljam can process and analyze SAM/BAM files in parallel and at high speed. The execution time with cljam is almost the same as with SAMtools. The cljam code is written in Clojure and has fewer lines than other similar tools.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
