{"title":"基于网络的富集基因子网络识别:一种博弈论方法。","authors":"Abolfazl Razi, Fatemeh Afghah, Salendra Singh, Vinay Varadan","doi":"10.4137/BECB.S38244","DOIUrl":null,"url":null,"abstract":"<p><p>Identifying subsets of genes that jointly mediate cancer etiology, progression, or therapy response remains a challenging problem due to the complexity and heterogeneity in cancer biology, a problem further exacerbated by the relatively small number of cancer samples profiled as compared with the sheer number of potential molecular factors involved. Pure data-driven methods that merely rely on multiomics data have been successful in discovering potentially functional genes but suffer from high false-positive rates and tend to report subsets of genes whose biological interrelationships are unclear. Recently, integrative data-driven models have been developed to integrate multiomics data with signaling pathway networks in order to identify pathways associated with clinical or biological phenotypes. However, these approaches suffer from an important drawback of being restricted to previously discovered pathway structures and miss novel genomic interactions as well as potential crosstalk among the pathways. In this article, we propose a novel coalition-based game-theoretic approach to overcome the challenge of identifying biologically relevant gene subnetworks associated with disease phenotypes. The algorithm starts from a set of seed genes and traverses a protein-protein interaction network to identify modulated subnetworks. The optimal set of modulated subnetworks is identified using Shapley value that accounts for both individual and collective utility of the subnetwork of genes. The algorithm is applied to two illustrative applications, including the identification of subnetworks associated with (i) disease progression risk in response to platinum-based therapy in ovarian cancer and (ii) immune infiltration in triple-negative breast cancer. The results demonstrate an improved predictive power of the proposed method when compared with state-of-the-art feature selection methods, with the added advantage of identifying novel potentially functional gene subnetworks that may provide insights into the mechanisms underlying cancer progression. </p>","PeriodicalId":42484,"journal":{"name":"Biomedical Engineering and Computational Biology","volume":"7 Suppl 2","pages":"1-14"},"PeriodicalIF":2.3000,"publicationDate":"2016-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4137/BECB.S38244","citationCount":"5","resultStr":"{\"title\":\"Network-Based Enriched Gene Subnetwork Identification: A Game-Theoretic Approach.\",\"authors\":\"Abolfazl Razi, Fatemeh Afghah, Salendra Singh, Vinay Varadan\",\"doi\":\"10.4137/BECB.S38244\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Identifying subsets of genes that jointly mediate cancer etiology, progression, or therapy response remains a challenging problem due to the complexity and heterogeneity in cancer biology, a problem further exacerbated by the relatively small number of cancer samples profiled as compared with the sheer number of potential molecular factors involved. Pure data-driven methods that merely rely on multiomics data have been successful in discovering potentially functional genes but suffer from high false-positive rates and tend to report subsets of genes whose biological interrelationships are unclear. Recently, integrative data-driven models have been developed to integrate multiomics data with signaling pathway networks in order to identify pathways associated with clinical or biological phenotypes. However, these approaches suffer from an important drawback of being restricted to previously discovered pathway structures and miss novel genomic interactions as well as potential crosstalk among the pathways. In this article, we propose a novel coalition-based game-theoretic approach to overcome the challenge of identifying biologically relevant gene subnetworks associated with disease phenotypes. The algorithm starts from a set of seed genes and traverses a protein-protein interaction network to identify modulated subnetworks. The optimal set of modulated subnetworks is identified using Shapley value that accounts for both individual and collective utility of the subnetwork of genes. The algorithm is applied to two illustrative applications, including the identification of subnetworks associated with (i) disease progression risk in response to platinum-based therapy in ovarian cancer and (ii) immune infiltration in triple-negative breast cancer. The results demonstrate an improved predictive power of the proposed method when compared with state-of-the-art feature selection methods, with the added advantage of identifying novel potentially functional gene subnetworks that may provide insights into the mechanisms underlying cancer progression. </p>\",\"PeriodicalId\":42484,\"journal\":{\"name\":\"Biomedical Engineering and Computational Biology\",\"volume\":\"7 Suppl 2\",\"pages\":\"1-14\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2016-04-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.4137/BECB.S38244\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomedical Engineering and Computational Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4137/BECB.S38244\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"ENGINEERING, BIOMEDICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomedical Engineering and Computational Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4137/BECB.S38244","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"ENGINEERING, BIOMEDICAL","Score":null,"Total":0}
Network-Based Enriched Gene Subnetwork Identification: A Game-Theoretic Approach.
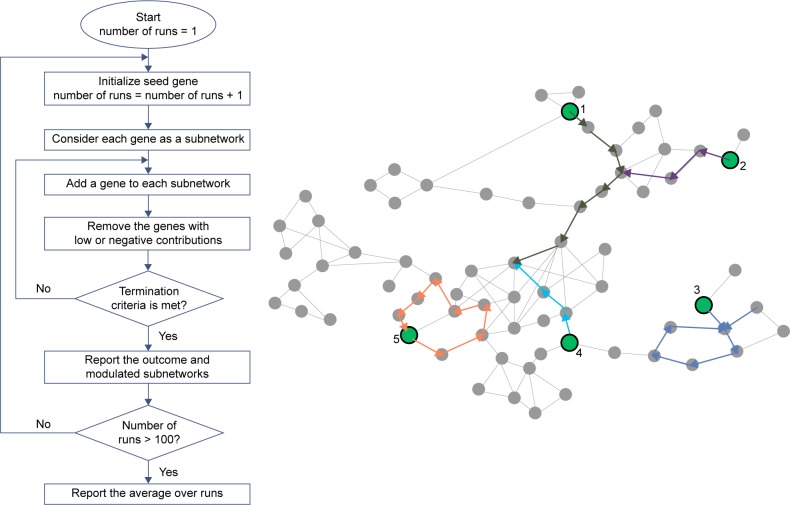
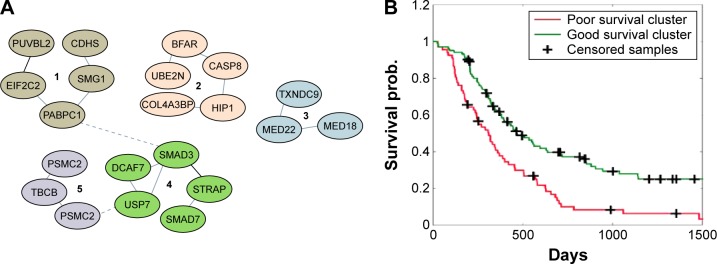
Identifying subsets of genes that jointly mediate cancer etiology, progression, or therapy response remains a challenging problem due to the complexity and heterogeneity in cancer biology, a problem further exacerbated by the relatively small number of cancer samples profiled as compared with the sheer number of potential molecular factors involved. Pure data-driven methods that merely rely on multiomics data have been successful in discovering potentially functional genes but suffer from high false-positive rates and tend to report subsets of genes whose biological interrelationships are unclear. Recently, integrative data-driven models have been developed to integrate multiomics data with signaling pathway networks in order to identify pathways associated with clinical or biological phenotypes. However, these approaches suffer from an important drawback of being restricted to previously discovered pathway structures and miss novel genomic interactions as well as potential crosstalk among the pathways. In this article, we propose a novel coalition-based game-theoretic approach to overcome the challenge of identifying biologically relevant gene subnetworks associated with disease phenotypes. The algorithm starts from a set of seed genes and traverses a protein-protein interaction network to identify modulated subnetworks. The optimal set of modulated subnetworks is identified using Shapley value that accounts for both individual and collective utility of the subnetwork of genes. The algorithm is applied to two illustrative applications, including the identification of subnetworks associated with (i) disease progression risk in response to platinum-based therapy in ovarian cancer and (ii) immune infiltration in triple-negative breast cancer. The results demonstrate an improved predictive power of the proposed method when compared with state-of-the-art feature selection methods, with the added advantage of identifying novel potentially functional gene subnetworks that may provide insights into the mechanisms underlying cancer progression.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
