Caleb F Davis, Deborah I Ritter, David A Wheeler, Hongmei Wang, Yan Ding, Shannon P Dugan, Matthew N Bainbridge, Donna M Muzny, Pulivarthi H Rao, Tsz-Kwong Man, Sharon E Plon, Richard A Gibbs, Ching C Lau
{"title":"SV-STAT通过对基于参考的组件进行校准来准确检测结构变化。","authors":"Caleb F Davis, Deborah I Ritter, David A Wheeler, Hongmei Wang, Yan Ding, Shannon P Dugan, Matthew N Bainbridge, Donna M Muzny, Pulivarthi H Rao, Tsz-Kwong Man, Sharon E Plon, Richard A Gibbs, Ching C Lau","doi":"10.1186/s13029-016-0051-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Genomic deletions, inversions, and other rearrangements known collectively as structural variations (SVs) are implicated in many human disorders. Technologies for sequencing DNA provide a potentially rich source of information in which to detect breakpoints of structural variations at base-pair resolution. However, accurate prediction of SVs remains challenging, and existing informatics tools predict rearrangements with significant rates of false positives or negatives.</p><p><strong>Results: </strong>To address this challenge, we developed 'Structural Variation detection by STAck and Tail' (SV-STAT) which implements a novel scoring metric. The software uses this statistic to quantify evidence for structural variation in genomic regions suspected of harboring rearrangements. To demonstrate SV-STAT, we used targeted and genome-wide approaches. First, we applied a custom capture array followed by Roche/454 and SV-STAT to three pediatric B-lineage acute lymphoblastic leukemias, identifying five structural variations joining known and novel breakpoint regions. Next, we detected SVs genome-wide in paired-end Illumina data collected from additional tumor samples. SV-STAT showed predictive accuracy as high as or higher than leading alternatives. The software is freely available under the terms of the GNU General Public License version 3 at https://gitorious.org/svstat/svstat.</p><p><strong>Conclusions: </strong>SV-STAT works across multiple sequencing chemistries, paired and single-end technologies, targeted or whole-genome strategies, and it complements existing SV-detection software. The method is a significant advance towards accurate detection and genotyping of genomic rearrangements from DNA sequencing data.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":" ","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2016-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-016-0051-0","citationCount":"4","resultStr":"{\"title\":\"SV-STAT accurately detects structural variation via alignment to reference-based assemblies.\",\"authors\":\"Caleb F Davis, Deborah I Ritter, David A Wheeler, Hongmei Wang, Yan Ding, Shannon P Dugan, Matthew N Bainbridge, Donna M Muzny, Pulivarthi H Rao, Tsz-Kwong Man, Sharon E Plon, Richard A Gibbs, Ching C Lau\",\"doi\":\"10.1186/s13029-016-0051-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Genomic deletions, inversions, and other rearrangements known collectively as structural variations (SVs) are implicated in many human disorders. Technologies for sequencing DNA provide a potentially rich source of information in which to detect breakpoints of structural variations at base-pair resolution. However, accurate prediction of SVs remains challenging, and existing informatics tools predict rearrangements with significant rates of false positives or negatives.</p><p><strong>Results: </strong>To address this challenge, we developed 'Structural Variation detection by STAck and Tail' (SV-STAT) which implements a novel scoring metric. The software uses this statistic to quantify evidence for structural variation in genomic regions suspected of harboring rearrangements. To demonstrate SV-STAT, we used targeted and genome-wide approaches. First, we applied a custom capture array followed by Roche/454 and SV-STAT to three pediatric B-lineage acute lymphoblastic leukemias, identifying five structural variations joining known and novel breakpoint regions. Next, we detected SVs genome-wide in paired-end Illumina data collected from additional tumor samples. SV-STAT showed predictive accuracy as high as or higher than leading alternatives. The software is freely available under the terms of the GNU General Public License version 3 at https://gitorious.org/svstat/svstat.</p><p><strong>Conclusions: </strong>SV-STAT works across multiple sequencing chemistries, paired and single-end technologies, targeted or whole-genome strategies, and it complements existing SV-detection software. The method is a significant advance towards accurate detection and genotyping of genomic rearrangements from DNA sequencing data.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\" \",\"pages\":\"8\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-016-0051-0\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-016-0051-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-016-0051-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
SV-STAT accurately detects structural variation via alignment to reference-based assemblies.
Background: Genomic deletions, inversions, and other rearrangements known collectively as structural variations (SVs) are implicated in many human disorders. Technologies for sequencing DNA provide a potentially rich source of information in which to detect breakpoints of structural variations at base-pair resolution. However, accurate prediction of SVs remains challenging, and existing informatics tools predict rearrangements with significant rates of false positives or negatives.
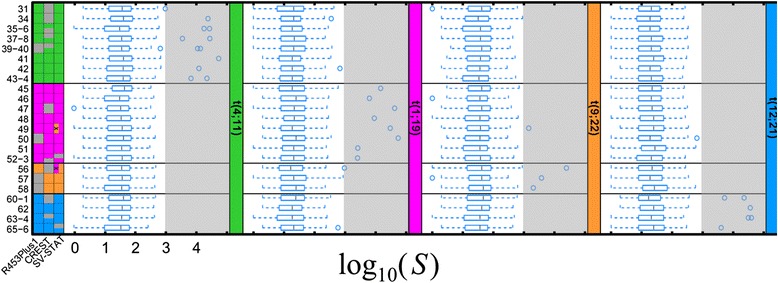
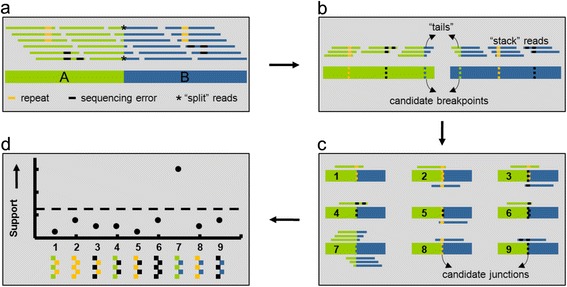
Results: To address this challenge, we developed 'Structural Variation detection by STAck and Tail' (SV-STAT) which implements a novel scoring metric. The software uses this statistic to quantify evidence for structural variation in genomic regions suspected of harboring rearrangements. To demonstrate SV-STAT, we used targeted and genome-wide approaches. First, we applied a custom capture array followed by Roche/454 and SV-STAT to three pediatric B-lineage acute lymphoblastic leukemias, identifying five structural variations joining known and novel breakpoint regions. Next, we detected SVs genome-wide in paired-end Illumina data collected from additional tumor samples. SV-STAT showed predictive accuracy as high as or higher than leading alternatives. The software is freely available under the terms of the GNU General Public License version 3 at https://gitorious.org/svstat/svstat.
Conclusions: SV-STAT works across multiple sequencing chemistries, paired and single-end technologies, targeted or whole-genome strategies, and it complements existing SV-detection software. The method is a significant advance towards accurate detection and genotyping of genomic rearrangements from DNA sequencing data.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
