{"title":"从全外显子组测序数据中排序序列变异的新工具。","authors":"Brigitte Glanzmann, Hendri Herbst, Craig J Kinnear, Marlo Möller, Junaid Gamieldien, Soraya Bardien","doi":"10.1186/s13029-016-0056-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Whole exome sequencing (WES) has provided a means for researchers to gain access to a highly enriched subset of the human genome in which to search for variants that are likely to be pathogenic and possibly provide important insights into disease mechanisms. In developing countries, bioinformatics capacity and expertise is severely limited and wet bench scientists are required to take on the challenging task of understanding and implementing the barrage of bioinformatics tools that are available to them.</p><p><strong>Results: </strong>We designed a novel method for the filtration of WES data called TAPER™ (Tool for Automated selection and Prioritization for Efficient Retrieval of sequence variants).</p><p><strong>Conclusions: </strong>TAPER™ implements a set of logical steps by which to prioritize candidate variants that could be associated with disease and this is aimed for implementation in biomedical laboratories with limited bioinformatics capacity. TAPER™ is free, can be setup on a Windows operating system (from Windows 7 and above) and does not require any programming knowledge. In summary, we have developed a freely available tool that simplifies variant prioritization from WES data in order to facilitate discovery of disease-causing genes.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":" ","pages":"10"},"PeriodicalIF":0.0000,"publicationDate":"2016-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-016-0056-8","citationCount":"8","resultStr":"{\"title\":\"A new tool for prioritization of sequence variants from whole exome sequencing data.\",\"authors\":\"Brigitte Glanzmann, Hendri Herbst, Craig J Kinnear, Marlo Möller, Junaid Gamieldien, Soraya Bardien\",\"doi\":\"10.1186/s13029-016-0056-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Whole exome sequencing (WES) has provided a means for researchers to gain access to a highly enriched subset of the human genome in which to search for variants that are likely to be pathogenic and possibly provide important insights into disease mechanisms. In developing countries, bioinformatics capacity and expertise is severely limited and wet bench scientists are required to take on the challenging task of understanding and implementing the barrage of bioinformatics tools that are available to them.</p><p><strong>Results: </strong>We designed a novel method for the filtration of WES data called TAPER™ (Tool for Automated selection and Prioritization for Efficient Retrieval of sequence variants).</p><p><strong>Conclusions: </strong>TAPER™ implements a set of logical steps by which to prioritize candidate variants that could be associated with disease and this is aimed for implementation in biomedical laboratories with limited bioinformatics capacity. TAPER™ is free, can be setup on a Windows operating system (from Windows 7 and above) and does not require any programming knowledge. In summary, we have developed a freely available tool that simplifies variant prioritization from WES data in order to facilitate discovery of disease-causing genes.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\" \",\"pages\":\"10\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-016-0056-8\",\"citationCount\":\"8\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-016-0056-8\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-016-0056-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
A new tool for prioritization of sequence variants from whole exome sequencing data.
Background: Whole exome sequencing (WES) has provided a means for researchers to gain access to a highly enriched subset of the human genome in which to search for variants that are likely to be pathogenic and possibly provide important insights into disease mechanisms. In developing countries, bioinformatics capacity and expertise is severely limited and wet bench scientists are required to take on the challenging task of understanding and implementing the barrage of bioinformatics tools that are available to them.
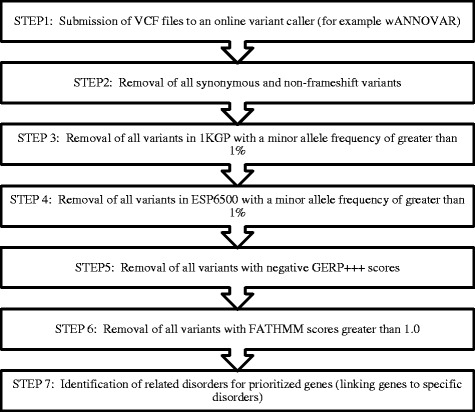
Results: We designed a novel method for the filtration of WES data called TAPER™ (Tool for Automated selection and Prioritization for Efficient Retrieval of sequence variants).
Conclusions: TAPER™ implements a set of logical steps by which to prioritize candidate variants that could be associated with disease and this is aimed for implementation in biomedical laboratories with limited bioinformatics capacity. TAPER™ is free, can be setup on a Windows operating system (from Windows 7 and above) and does not require any programming knowledge. In summary, we have developed a freely available tool that simplifies variant prioritization from WES data in order to facilitate discovery of disease-causing genes.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
