{"title":"TagDigger:用户友好的从GBS和RAD-seq数据中提取读取计数。","authors":"Lindsay V Clark, Erik J Sacks","doi":"10.1186/s13029-016-0057-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In genotyping-by-sequencing (GBS) and restriction site-associated DNA sequencing (RAD-seq), read depth is important for assessing the quality of genotype calls and estimating allele dosage in polyploids. However, existing pipelines for GBS and RAD-seq do not provide read counts in formats that are both accurate and easy to access. Additionally, although existing pipelines allow previously-mined SNPs to be genotyped on new samples, they do not allow the user to manually specify a subset of loci to examine. Pipelines that do not use a reference genome assign arbitrary names to SNPs, making meta-analysis across projects difficult.</p><p><strong>Results: </strong>We created the software TagDigger, which includes three programs for analyzing GBS and RAD-seq data. The first script, tagdigger_interactive.py, rapidly extracts read counts and genotypes from FASTQ files using user-supplied sets of barcodes and tags. Input and output is in CSV format so that it can be opened by spreadsheet software. Tag sequences can also be imported from the Stacks, TASSEL-GBSv2, TASSEL-UNEAK, or pyRAD pipelines, and a separate file can be imported listing the names of markers to retain. A second script, tag_manager.py, consolidates marker names and sequences across multiple projects. A third script, barcode_splitter.py, assists with preparing FASTQ data for deposit in a public archive by splitting FASTQ files by barcode and generating MD5 checksums for the resulting files.</p><p><strong>Conclusions: </strong>TagDigger is open-source and freely available software written in Python 3. It uses a scalable, rapid search algorithm that can process over 100 million FASTQ reads per hour. TagDigger will run on a laptop with any operating system, does not consume hard drive space with intermediate files, and does not require programming skill to use.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":" ","pages":"11"},"PeriodicalIF":0.0000,"publicationDate":"2016-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-016-0057-7","citationCount":"11","resultStr":"{\"title\":\"TagDigger: user-friendly extraction of read counts from GBS and RAD-seq data.\",\"authors\":\"Lindsay V Clark, Erik J Sacks\",\"doi\":\"10.1186/s13029-016-0057-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>In genotyping-by-sequencing (GBS) and restriction site-associated DNA sequencing (RAD-seq), read depth is important for assessing the quality of genotype calls and estimating allele dosage in polyploids. However, existing pipelines for GBS and RAD-seq do not provide read counts in formats that are both accurate and easy to access. Additionally, although existing pipelines allow previously-mined SNPs to be genotyped on new samples, they do not allow the user to manually specify a subset of loci to examine. Pipelines that do not use a reference genome assign arbitrary names to SNPs, making meta-analysis across projects difficult.</p><p><strong>Results: </strong>We created the software TagDigger, which includes three programs for analyzing GBS and RAD-seq data. The first script, tagdigger_interactive.py, rapidly extracts read counts and genotypes from FASTQ files using user-supplied sets of barcodes and tags. Input and output is in CSV format so that it can be opened by spreadsheet software. Tag sequences can also be imported from the Stacks, TASSEL-GBSv2, TASSEL-UNEAK, or pyRAD pipelines, and a separate file can be imported listing the names of markers to retain. A second script, tag_manager.py, consolidates marker names and sequences across multiple projects. A third script, barcode_splitter.py, assists with preparing FASTQ data for deposit in a public archive by splitting FASTQ files by barcode and generating MD5 checksums for the resulting files.</p><p><strong>Conclusions: </strong>TagDigger is open-source and freely available software written in Python 3. It uses a scalable, rapid search algorithm that can process over 100 million FASTQ reads per hour. TagDigger will run on a laptop with any operating system, does not consume hard drive space with intermediate files, and does not require programming skill to use.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\" \",\"pages\":\"11\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-07-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-016-0057-7\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-016-0057-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-016-0057-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
TagDigger: user-friendly extraction of read counts from GBS and RAD-seq data.
Background: In genotyping-by-sequencing (GBS) and restriction site-associated DNA sequencing (RAD-seq), read depth is important for assessing the quality of genotype calls and estimating allele dosage in polyploids. However, existing pipelines for GBS and RAD-seq do not provide read counts in formats that are both accurate and easy to access. Additionally, although existing pipelines allow previously-mined SNPs to be genotyped on new samples, they do not allow the user to manually specify a subset of loci to examine. Pipelines that do not use a reference genome assign arbitrary names to SNPs, making meta-analysis across projects difficult.
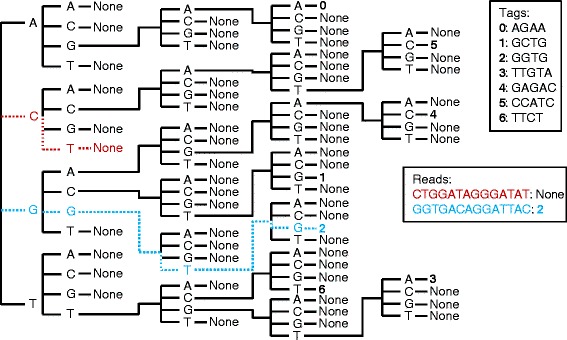
Results: We created the software TagDigger, which includes three programs for analyzing GBS and RAD-seq data. The first script, tagdigger_interactive.py, rapidly extracts read counts and genotypes from FASTQ files using user-supplied sets of barcodes and tags. Input and output is in CSV format so that it can be opened by spreadsheet software. Tag sequences can also be imported from the Stacks, TASSEL-GBSv2, TASSEL-UNEAK, or pyRAD pipelines, and a separate file can be imported listing the names of markers to retain. A second script, tag_manager.py, consolidates marker names and sequences across multiple projects. A third script, barcode_splitter.py, assists with preparing FASTQ data for deposit in a public archive by splitting FASTQ files by barcode and generating MD5 checksums for the resulting files.
Conclusions: TagDigger is open-source and freely available software written in Python 3. It uses a scalable, rapid search algorithm that can process over 100 million FASTQ reads per hour. TagDigger will run on a laptop with any operating system, does not consume hard drive space with intermediate files, and does not require programming skill to use.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
