Masharip Atadzhanov, Danielle C Smith, Mwila H Mwaba, Omar K Siddiqi, Alan Bryer, L Jacquie Greenberg
{"title":"赞比亚家族脊髓小脑性共济失调7型(SCA7)的临床和遗传分析。","authors":"Masharip Atadzhanov, Danielle C Smith, Mwila H Mwaba, Omar K Siddiqi, Alan Bryer, L Jacquie Greenberg","doi":"10.1186/s40673-017-0075-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>To date, 43 types of Spinocerebellar Ataxias (SCAs) have been identified. A subset of the SCAs are caused by the pathogenic expansion of a CAG repeat tract within the corresponding gene. Ethnic and geographic differences are evident in the prevalence of the autosomal dominant SCAs. Few descriptions of the clinical phenotype and molecular genetics of the SCAs are available from the African continent. Established studies mostly concern the South African populations, where there is a high frequency of SCA1, SCA2 and SCA7. The SCA7 mutation in South Africa (SA) has been found almost exclusively in families of indigenous Black African ethnic origin.</p><p><strong>Objective: </strong>To present the results of the first clinical description of seven Zambian families presenting with autosomal dominant SCA, as well as the downstream molecular genetic analysis of a subset of these families.</p><p><strong>Methods: </strong>The study was undertaken at the University Teaching Hospital in Lusaka, Zambia. Ataxia was quantified with the Brief Ataxia Rating Scale derived from the modified international ataxia rating scale. Molecular genetic testing for 5 types of SCA (SCA1, SCA2, SCA3, SCA6 and SCA7) was performed at the National Health Laboratory Service at Groote Schuur Hospital and the Division of Human Genetics, University of Cape Town, SA. The clinical and radiological features were evaluated in seven families with autosomal dominant cerebellar ataxia. Molecular genetic analysis was completed on individuals representing three of the seven families.</p><p><strong>Results: </strong>All affected families were ethnic Zambians from various tribes, originating from three different regions of the country (Eastern, Western and Central province). Thirty-four individuals from four families had phenotypic features of SCA7. SCA7 was confirmed by molecular testing in 10 individuals from 3 of these families. The age of onset of the disease varied from 12 to 59 years. The most prominent phenotypic features in these families were gait and limb ataxia, dysarthria, visual loss, ptosis, ophthalmoparesis/ophthalmoplegia, pyramidal tract signs, and dementia. Affected members of the SCA7 families had progressive macular degeneration and cerebellar atrophy. All families displayed marked anticipation of age at onset and rate of symptom progression. The pathogenic SCA7 CAG repeat ranges varied from 47 to 56 repeats. Three additional families were found to have clinical phenotypes associated with autosomal dominant SCA, however, DNA was not available for molecular confirmation. The age of onset of the disease in these families varied from 19 to 53 years. The most common clinical picture in these families included a combination of cerebellar symptoms with slow saccadic eye movements, peripheral neuropathy, dementia and tremor.</p><p><strong>Conclusion: </strong>SCA is prevalent in ethnic Zambian families. The SCA7 families in this report had similar clinical presentations to families described in other African countries. In all families, the disease had an autosomal dominant pattern of inheritance across multiple generations. All families displayed anticipation of both age of onset and the rate of disease progression. Further clinical and molecular investigations of the inherited ataxias in a larger cohort of patients is important to understand the natural history and origin of SCAs in the Zambian population.</p>","PeriodicalId":36752,"journal":{"name":"Cerebellum and Ataxias","volume":"4 ","pages":"17"},"PeriodicalIF":0.0000,"publicationDate":"2017-11-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40673-017-0075-5","citationCount":"7","resultStr":"{\"title\":\"Clinical and genetic analysis of spinocerebellar ataxia type 7 (SCA7) in Zambian families.\",\"authors\":\"Masharip Atadzhanov, Danielle C Smith, Mwila H Mwaba, Omar K Siddiqi, Alan Bryer, L Jacquie Greenberg\",\"doi\":\"10.1186/s40673-017-0075-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>To date, 43 types of Spinocerebellar Ataxias (SCAs) have been identified. A subset of the SCAs are caused by the pathogenic expansion of a CAG repeat tract within the corresponding gene. Ethnic and geographic differences are evident in the prevalence of the autosomal dominant SCAs. Few descriptions of the clinical phenotype and molecular genetics of the SCAs are available from the African continent. Established studies mostly concern the South African populations, where there is a high frequency of SCA1, SCA2 and SCA7. The SCA7 mutation in South Africa (SA) has been found almost exclusively in families of indigenous Black African ethnic origin.</p><p><strong>Objective: </strong>To present the results of the first clinical description of seven Zambian families presenting with autosomal dominant SCA, as well as the downstream molecular genetic analysis of a subset of these families.</p><p><strong>Methods: </strong>The study was undertaken at the University Teaching Hospital in Lusaka, Zambia. Ataxia was quantified with the Brief Ataxia Rating Scale derived from the modified international ataxia rating scale. Molecular genetic testing for 5 types of SCA (SCA1, SCA2, SCA3, SCA6 and SCA7) was performed at the National Health Laboratory Service at Groote Schuur Hospital and the Division of Human Genetics, University of Cape Town, SA. The clinical and radiological features were evaluated in seven families with autosomal dominant cerebellar ataxia. Molecular genetic analysis was completed on individuals representing three of the seven families.</p><p><strong>Results: </strong>All affected families were ethnic Zambians from various tribes, originating from three different regions of the country (Eastern, Western and Central province). Thirty-four individuals from four families had phenotypic features of SCA7. SCA7 was confirmed by molecular testing in 10 individuals from 3 of these families. The age of onset of the disease varied from 12 to 59 years. The most prominent phenotypic features in these families were gait and limb ataxia, dysarthria, visual loss, ptosis, ophthalmoparesis/ophthalmoplegia, pyramidal tract signs, and dementia. Affected members of the SCA7 families had progressive macular degeneration and cerebellar atrophy. All families displayed marked anticipation of age at onset and rate of symptom progression. The pathogenic SCA7 CAG repeat ranges varied from 47 to 56 repeats. Three additional families were found to have clinical phenotypes associated with autosomal dominant SCA, however, DNA was not available for molecular confirmation. The age of onset of the disease in these families varied from 19 to 53 years. The most common clinical picture in these families included a combination of cerebellar symptoms with slow saccadic eye movements, peripheral neuropathy, dementia and tremor.</p><p><strong>Conclusion: </strong>SCA is prevalent in ethnic Zambian families. The SCA7 families in this report had similar clinical presentations to families described in other African countries. In all families, the disease had an autosomal dominant pattern of inheritance across multiple generations. All families displayed anticipation of both age of onset and the rate of disease progression. Further clinical and molecular investigations of the inherited ataxias in a larger cohort of patients is important to understand the natural history and origin of SCAs in the Zambian population.</p>\",\"PeriodicalId\":36752,\"journal\":{\"name\":\"Cerebellum and Ataxias\",\"volume\":\"4 \",\"pages\":\"17\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2017-11-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s40673-017-0075-5\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cerebellum and Ataxias\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40673-017-0075-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2017/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cerebellum and Ataxias","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40673-017-0075-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
Clinical and genetic analysis of spinocerebellar ataxia type 7 (SCA7) in Zambian families.
Background: To date, 43 types of Spinocerebellar Ataxias (SCAs) have been identified. A subset of the SCAs are caused by the pathogenic expansion of a CAG repeat tract within the corresponding gene. Ethnic and geographic differences are evident in the prevalence of the autosomal dominant SCAs. Few descriptions of the clinical phenotype and molecular genetics of the SCAs are available from the African continent. Established studies mostly concern the South African populations, where there is a high frequency of SCA1, SCA2 and SCA7. The SCA7 mutation in South Africa (SA) has been found almost exclusively in families of indigenous Black African ethnic origin.
Objective: To present the results of the first clinical description of seven Zambian families presenting with autosomal dominant SCA, as well as the downstream molecular genetic analysis of a subset of these families.
Methods: The study was undertaken at the University Teaching Hospital in Lusaka, Zambia. Ataxia was quantified with the Brief Ataxia Rating Scale derived from the modified international ataxia rating scale. Molecular genetic testing for 5 types of SCA (SCA1, SCA2, SCA3, SCA6 and SCA7) was performed at the National Health Laboratory Service at Groote Schuur Hospital and the Division of Human Genetics, University of Cape Town, SA. The clinical and radiological features were evaluated in seven families with autosomal dominant cerebellar ataxia. Molecular genetic analysis was completed on individuals representing three of the seven families.
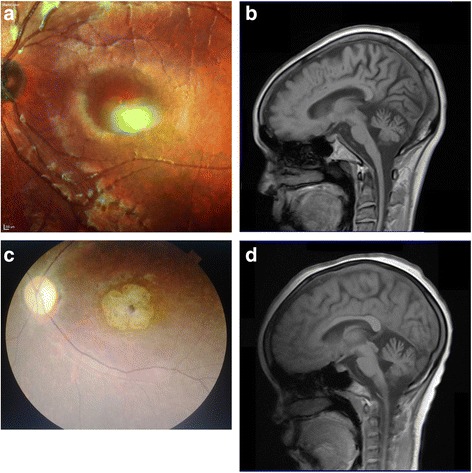
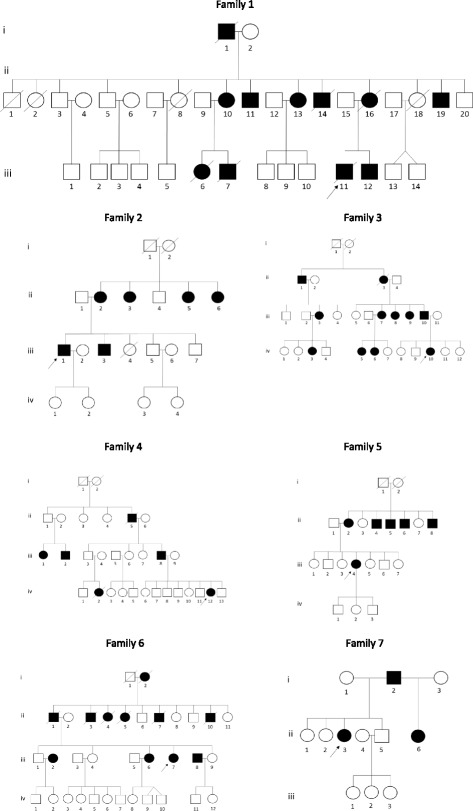
Results: All affected families were ethnic Zambians from various tribes, originating from three different regions of the country (Eastern, Western and Central province). Thirty-four individuals from four families had phenotypic features of SCA7. SCA7 was confirmed by molecular testing in 10 individuals from 3 of these families. The age of onset of the disease varied from 12 to 59 years. The most prominent phenotypic features in these families were gait and limb ataxia, dysarthria, visual loss, ptosis, ophthalmoparesis/ophthalmoplegia, pyramidal tract signs, and dementia. Affected members of the SCA7 families had progressive macular degeneration and cerebellar atrophy. All families displayed marked anticipation of age at onset and rate of symptom progression. The pathogenic SCA7 CAG repeat ranges varied from 47 to 56 repeats. Three additional families were found to have clinical phenotypes associated with autosomal dominant SCA, however, DNA was not available for molecular confirmation. The age of onset of the disease in these families varied from 19 to 53 years. The most common clinical picture in these families included a combination of cerebellar symptoms with slow saccadic eye movements, peripheral neuropathy, dementia and tremor.
Conclusion: SCA is prevalent in ethnic Zambian families. The SCA7 families in this report had similar clinical presentations to families described in other African countries. In all families, the disease had an autosomal dominant pattern of inheritance across multiple generations. All families displayed anticipation of both age of onset and the rate of disease progression. Further clinical and molecular investigations of the inherited ataxias in a larger cohort of patients is important to understand the natural history and origin of SCAs in the Zambian population.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
