{"title":"SOV_refine:进一步细化了片段重叠评分的定义及其对蛋白质结构相似性的意义。","authors":"Tong Liu, Zheng Wang","doi":"10.1186/s13029-018-0068-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The segment overlap score (SOV) has been used to evaluate the predicted protein secondary structures, a sequence composed of helix (H), strand (E), and coil (C), by comparing it with the native or reference secondary structures, another sequence of H, E, and C. SOV's advantage is that it can consider the size of continuous overlapping segments and assign extra allowance to longer continuous overlapping segments instead of only judging from the percentage of overlapping individual positions as Q3 score does. However, we have found a drawback from its previous definition, that is, it cannot ensure increasing allowance assignment when more residues in a segment are further predicted accurately.</p><p><strong>Results: </strong>A new way of assigning allowance has been designed, which keeps all the advantages of the previous SOV score definitions and ensures that the amount of allowance assigned is incremental when more elements in a segment are predicted accurately. Furthermore, our improved SOV has achieved a higher correlation with the quality of protein models measured by GDT-TS score and TM-score, indicating its better abilities to evaluate tertiary structure quality at the secondary structure level. We analyzed the statistical significance of SOV scores and found the threshold values for distinguishing two protein structures (SOV_refine > 0.19) and indicating whether two proteins are under the same CATH fold (SOV_refine > 0.94 and > 0.90 for three- and eight-state secondary structures respectively). We provided another two example applications, which are when used as a machine learning feature for protein model quality assessment and comparing different definitions of topologically associating domains. We proved that our newly defined SOV score resulted in better performance.</p><p><strong>Conclusions: </strong>The SOV score can be widely used in bioinformatics research and other fields that need to compare two sequences of letters in which continuous segments have important meanings. We also generalized the previous SOV definitions so that it can work for sequences composed of more than three states (e.g., it can work for the eight-state definition of protein secondary structures). A standalone software package has been implemented in Perl with source code released. The software can be downloaded from http://dna.cs.miami.edu/SOV/.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":"13 ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2018-04-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-018-0068-7","citationCount":"16","resultStr":"{\"title\":\"SOV_refine: A further refined definition of segment overlap score and its significance for protein structure similarity.\",\"authors\":\"Tong Liu, Zheng Wang\",\"doi\":\"10.1186/s13029-018-0068-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The segment overlap score (SOV) has been used to evaluate the predicted protein secondary structures, a sequence composed of helix (H), strand (E), and coil (C), by comparing it with the native or reference secondary structures, another sequence of H, E, and C. SOV's advantage is that it can consider the size of continuous overlapping segments and assign extra allowance to longer continuous overlapping segments instead of only judging from the percentage of overlapping individual positions as Q3 score does. However, we have found a drawback from its previous definition, that is, it cannot ensure increasing allowance assignment when more residues in a segment are further predicted accurately.</p><p><strong>Results: </strong>A new way of assigning allowance has been designed, which keeps all the advantages of the previous SOV score definitions and ensures that the amount of allowance assigned is incremental when more elements in a segment are predicted accurately. Furthermore, our improved SOV has achieved a higher correlation with the quality of protein models measured by GDT-TS score and TM-score, indicating its better abilities to evaluate tertiary structure quality at the secondary structure level. We analyzed the statistical significance of SOV scores and found the threshold values for distinguishing two protein structures (SOV_refine > 0.19) and indicating whether two proteins are under the same CATH fold (SOV_refine > 0.94 and > 0.90 for three- and eight-state secondary structures respectively). We provided another two example applications, which are when used as a machine learning feature for protein model quality assessment and comparing different definitions of topologically associating domains. We proved that our newly defined SOV score resulted in better performance.</p><p><strong>Conclusions: </strong>The SOV score can be widely used in bioinformatics research and other fields that need to compare two sequences of letters in which continuous segments have important meanings. We also generalized the previous SOV definitions so that it can work for sequences composed of more than three states (e.g., it can work for the eight-state definition of protein secondary structures). A standalone software package has been implemented in Perl with source code released. The software can be downloaded from http://dna.cs.miami.edu/SOV/.</p>\",\"PeriodicalId\":35052,\"journal\":{\"name\":\"Source Code for Biology and Medicine\",\"volume\":\"13 \",\"pages\":\"1\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-04-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13029-018-0068-7\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Source Code for Biology and Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13029-018-0068-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Decision Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-018-0068-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
SOV_refine: A further refined definition of segment overlap score and its significance for protein structure similarity.
Background: The segment overlap score (SOV) has been used to evaluate the predicted protein secondary structures, a sequence composed of helix (H), strand (E), and coil (C), by comparing it with the native or reference secondary structures, another sequence of H, E, and C. SOV's advantage is that it can consider the size of continuous overlapping segments and assign extra allowance to longer continuous overlapping segments instead of only judging from the percentage of overlapping individual positions as Q3 score does. However, we have found a drawback from its previous definition, that is, it cannot ensure increasing allowance assignment when more residues in a segment are further predicted accurately.
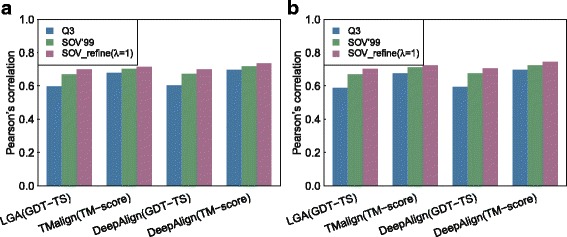
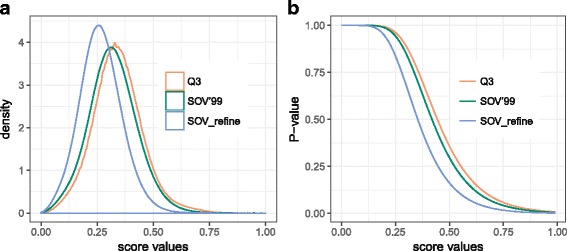
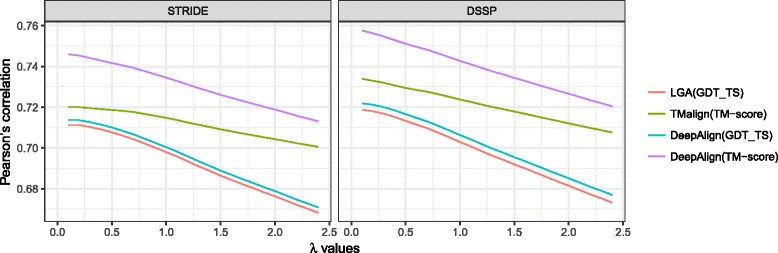
Results: A new way of assigning allowance has been designed, which keeps all the advantages of the previous SOV score definitions and ensures that the amount of allowance assigned is incremental when more elements in a segment are predicted accurately. Furthermore, our improved SOV has achieved a higher correlation with the quality of protein models measured by GDT-TS score and TM-score, indicating its better abilities to evaluate tertiary structure quality at the secondary structure level. We analyzed the statistical significance of SOV scores and found the threshold values for distinguishing two protein structures (SOV_refine > 0.19) and indicating whether two proteins are under the same CATH fold (SOV_refine > 0.94 and > 0.90 for three- and eight-state secondary structures respectively). We provided another two example applications, which are when used as a machine learning feature for protein model quality assessment and comparing different definitions of topologically associating domains. We proved that our newly defined SOV score resulted in better performance.
Conclusions: The SOV score can be widely used in bioinformatics research and other fields that need to compare two sequences of letters in which continuous segments have important meanings. We also generalized the previous SOV definitions so that it can work for sequences composed of more than three states (e.g., it can work for the eight-state definition of protein secondary structures). A standalone software package has been implemented in Perl with source code released. The software can be downloaded from http://dna.cs.miami.edu/SOV/.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
