Yu. Kondrakhin , T. Valeev , R. Sharipov , I. Yevshin , F. Kolpakov , A. Kel
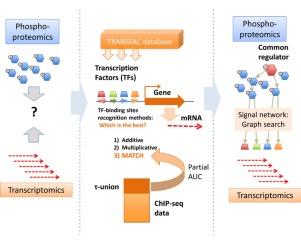
{"title":"结合蛋白质组学和转录组学数据的转录因子的蛋白质- dna相互作用预测","authors":"Yu. Kondrakhin , T. Valeev , R. Sharipov , I. Yevshin , F. Kolpakov , A. Kel","doi":"10.1016/j.euprot.2016.09.001","DOIUrl":null,"url":null,"abstract":"<div><p>We compared positional weight matrix-based prediction methods for transcription factor (TF) binding sites using selected fraction of ChIP-seq data with the help of partial AUC measure (limited to false positive rate 0.1, that is the most relevant for the application of the TF search in the genome scale). Comparison of three prediction methods—additive, multiplicative and information-vector based (MATCH) showed an advantage of the MATCH method for majority of transcription factors tested. We demonstrated that application of TF site identifying methods can help to connect the proteomics and phosphoproteomics world of signaling networks to gene regulation and transcriptomics world.</p></div>","PeriodicalId":38260,"journal":{"name":"EuPA Open Proteomics","volume":"13 ","pages":"Pages 14-23"},"PeriodicalIF":0.0000,"publicationDate":"2016-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.euprot.2016.09.001","citationCount":"4","resultStr":"{\"title\":\"Prediction of protein-DNA interactions of transcription factors linking proteomics and transcriptomics data\",\"authors\":\"Yu. Kondrakhin , T. Valeev , R. Sharipov , I. Yevshin , F. Kolpakov , A. Kel\",\"doi\":\"10.1016/j.euprot.2016.09.001\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>We compared positional weight matrix-based prediction methods for transcription factor (TF) binding sites using selected fraction of ChIP-seq data with the help of partial AUC measure (limited to false positive rate 0.1, that is the most relevant for the application of the TF search in the genome scale). Comparison of three prediction methods—additive, multiplicative and information-vector based (MATCH) showed an advantage of the MATCH method for majority of transcription factors tested. We demonstrated that application of TF site identifying methods can help to connect the proteomics and phosphoproteomics world of signaling networks to gene regulation and transcriptomics world.</p></div>\",\"PeriodicalId\":38260,\"journal\":{\"name\":\"EuPA Open Proteomics\",\"volume\":\"13 \",\"pages\":\"Pages 14-23\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.euprot.2016.09.001\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EuPA Open Proteomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2212968516300447\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/9/15 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EuPA Open Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2212968516300447","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/9/15 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Prediction of protein-DNA interactions of transcription factors linking proteomics and transcriptomics data
We compared positional weight matrix-based prediction methods for transcription factor (TF) binding sites using selected fraction of ChIP-seq data with the help of partial AUC measure (limited to false positive rate 0.1, that is the most relevant for the application of the TF search in the genome scale). Comparison of three prediction methods—additive, multiplicative and information-vector based (MATCH) showed an advantage of the MATCH method for majority of transcription factors tested. We demonstrated that application of TF site identifying methods can help to connect the proteomics and phosphoproteomics world of signaling networks to gene regulation and transcriptomics world.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
