{"title":"阿苯达唑、三唑苯达唑、涕灭威、甲氧威、孟鲁司特和齐拉西酮的亚砜和/或砜类代谢物的体外药物相互作用潜能。","authors":"Poonam Giri, Lakshmikant Gupta, Sneha Naidu, Vipul Joshi, Nirmal Patel, Shyamkumar Giri, Nuggehally R Srinivas","doi":"10.2174/1872312812666180816164626","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The use of polypharmacy in the present day clinical therapy has made the identification of clinical drug-drug interaction risk an important aspect of drug development process. Although many drugs can be metabolized to sulfoxide and/or sulfone metabolites, seldom is known on the CYP inhibition potential and/or the metabolic fate for such metabolites.</p><p><strong>Objective: </strong>The key objectives were: a) to evaluate the in vitro CYP inhibition potential of selected parent drugs with sulfoxide/sulfone metabolites; b) to assess the in vitro metabolic fate of the same panel of parent drugs and metabolites.</p><p><strong>Methods: </strong>In vitro drug-drug interaction potential of test compounds was investigated in two stages; 1) assessment of CYP450 inhibition potential of test compounds using human liver microsomes (HLM); and 2) assessment of test compounds as substrate of Phase I enzymes; including CYP450, FMO, AO and MAO using HLM, recombinant human CYP enzymes (rhCYP), Human Liver Cytosol (HLC) and Human Liver Mitochondrial (HLMit). All samples were analysed by LC-MS-MS method.</p><p><strong>Results: </strong>CYP1A2 was inhibited by methiocarb, triclabendazole, triclabendazole sulfoxide, and ziprasidone sulfone with IC50 of 0.71 µM, 1.07 µM, 4.19 µM, and 17.14 µM, respectively. CYP2C8 was inhibited by montelukast, montelukast sulfoxide, montelukast sulfone, tribendazole, triclabendazole sulfoxide, and triclabendazole sulfone with IC50 of 0.08 µM, 0.05 µM, 0.02 µM, 3.31 µM, 8.95 µM, and 1.05 µM, respectively. CYP2C9 was inhibited by triclabendazole, triclabendazole sulfoxide, triclabendazole sulfone, montelukast, montelukast sulfoxide and montelukast sulfone with IC50 of 1.17 µM, 1.95 µM, 0.69 µM, 1.34 µM, 3.61 µM and 2.15 µM, respectively. CYP2C19 was inhibited by triclabendazole and triclabendazole sulfoxide with IC50 of 0.25 and 0.22, respectively. CYP3A4 was inhibited by montelukast sulfoxide and triclabendazole with IC50 of 9.33 and 15.11, respectively. Amongst the studied sulfoxide/sulfone substrates, the propensity of involvement of CY2C9 and CYP3A4 enzyme was high (approximately 56% of total) in the metabolic fate experiments.</p><p><strong>Conclusion: </strong>Based on the findings, a proper risk assessment strategy needs to be factored (i.e., perpetrator and/or victim drug) to overcome any imminent risk of potential clinical drug-drug interaction when sulfoxide/sulfone metabolite(s) generating drugs are coadministered in therapy.</p>","PeriodicalId":11339,"journal":{"name":"Drug metabolism letters","volume":" ","pages":"101-116"},"PeriodicalIF":0.0000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/51/7d/DML-12-101.PMC6416464.pdf","citationCount":"0","resultStr":"{\"title\":\"In Vitro Drug-Drug Interaction Potential of Sulfoxide and/or Sulfone Metabolites of Albendazole, Triclabendazole , Aldicarb, Methiocarb, Montelukast and Ziprasidone.\",\"authors\":\"Poonam Giri, Lakshmikant Gupta, Sneha Naidu, Vipul Joshi, Nirmal Patel, Shyamkumar Giri, Nuggehally R Srinivas\",\"doi\":\"10.2174/1872312812666180816164626\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The use of polypharmacy in the present day clinical therapy has made the identification of clinical drug-drug interaction risk an important aspect of drug development process. Although many drugs can be metabolized to sulfoxide and/or sulfone metabolites, seldom is known on the CYP inhibition potential and/or the metabolic fate for such metabolites.</p><p><strong>Objective: </strong>The key objectives were: a) to evaluate the in vitro CYP inhibition potential of selected parent drugs with sulfoxide/sulfone metabolites; b) to assess the in vitro metabolic fate of the same panel of parent drugs and metabolites.</p><p><strong>Methods: </strong>In vitro drug-drug interaction potential of test compounds was investigated in two stages; 1) assessment of CYP450 inhibition potential of test compounds using human liver microsomes (HLM); and 2) assessment of test compounds as substrate of Phase I enzymes; including CYP450, FMO, AO and MAO using HLM, recombinant human CYP enzymes (rhCYP), Human Liver Cytosol (HLC) and Human Liver Mitochondrial (HLMit). All samples were analysed by LC-MS-MS method.</p><p><strong>Results: </strong>CYP1A2 was inhibited by methiocarb, triclabendazole, triclabendazole sulfoxide, and ziprasidone sulfone with IC50 of 0.71 µM, 1.07 µM, 4.19 µM, and 17.14 µM, respectively. CYP2C8 was inhibited by montelukast, montelukast sulfoxide, montelukast sulfone, tribendazole, triclabendazole sulfoxide, and triclabendazole sulfone with IC50 of 0.08 µM, 0.05 µM, 0.02 µM, 3.31 µM, 8.95 µM, and 1.05 µM, respectively. CYP2C9 was inhibited by triclabendazole, triclabendazole sulfoxide, triclabendazole sulfone, montelukast, montelukast sulfoxide and montelukast sulfone with IC50 of 1.17 µM, 1.95 µM, 0.69 µM, 1.34 µM, 3.61 µM and 2.15 µM, respectively. CYP2C19 was inhibited by triclabendazole and triclabendazole sulfoxide with IC50 of 0.25 and 0.22, respectively. CYP3A4 was inhibited by montelukast sulfoxide and triclabendazole with IC50 of 9.33 and 15.11, respectively. Amongst the studied sulfoxide/sulfone substrates, the propensity of involvement of CY2C9 and CYP3A4 enzyme was high (approximately 56% of total) in the metabolic fate experiments.</p><p><strong>Conclusion: </strong>Based on the findings, a proper risk assessment strategy needs to be factored (i.e., perpetrator and/or victim drug) to overcome any imminent risk of potential clinical drug-drug interaction when sulfoxide/sulfone metabolite(s) generating drugs are coadministered in therapy.</p>\",\"PeriodicalId\":11339,\"journal\":{\"name\":\"Drug metabolism letters\",\"volume\":\" \",\"pages\":\"101-116\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/51/7d/DML-12-101.PMC6416464.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Drug metabolism letters\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2174/1872312812666180816164626\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Drug metabolism letters","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2174/1872312812666180816164626","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
In Vitro Drug-Drug Interaction Potential of Sulfoxide and/or Sulfone Metabolites of Albendazole, Triclabendazole , Aldicarb, Methiocarb, Montelukast and Ziprasidone.
Background: The use of polypharmacy in the present day clinical therapy has made the identification of clinical drug-drug interaction risk an important aspect of drug development process. Although many drugs can be metabolized to sulfoxide and/or sulfone metabolites, seldom is known on the CYP inhibition potential and/or the metabolic fate for such metabolites.
Objective: The key objectives were: a) to evaluate the in vitro CYP inhibition potential of selected parent drugs with sulfoxide/sulfone metabolites; b) to assess the in vitro metabolic fate of the same panel of parent drugs and metabolites.
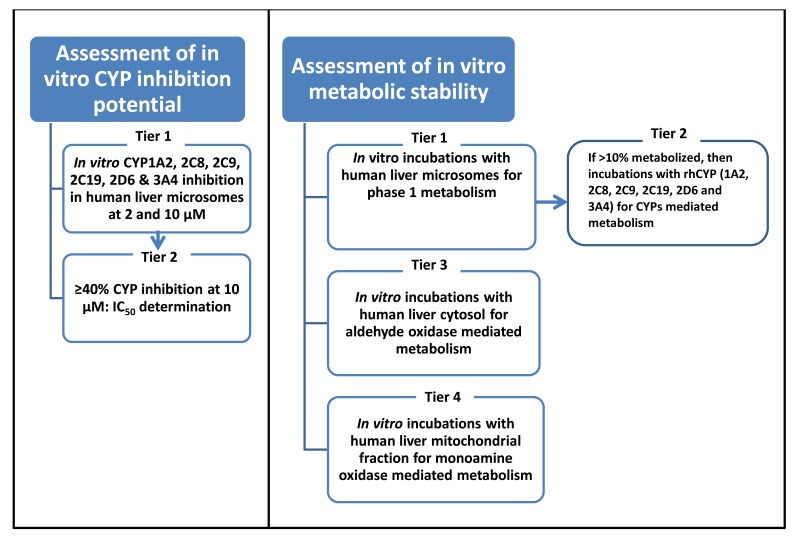
Methods: In vitro drug-drug interaction potential of test compounds was investigated in two stages; 1) assessment of CYP450 inhibition potential of test compounds using human liver microsomes (HLM); and 2) assessment of test compounds as substrate of Phase I enzymes; including CYP450, FMO, AO and MAO using HLM, recombinant human CYP enzymes (rhCYP), Human Liver Cytosol (HLC) and Human Liver Mitochondrial (HLMit). All samples were analysed by LC-MS-MS method.
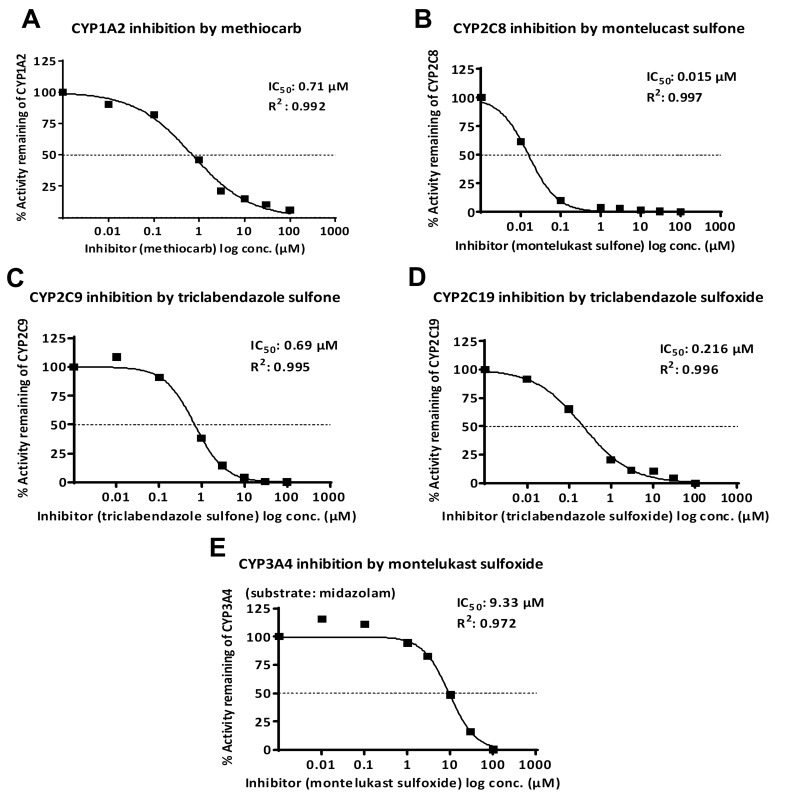
Results: CYP1A2 was inhibited by methiocarb, triclabendazole, triclabendazole sulfoxide, and ziprasidone sulfone with IC50 of 0.71 µM, 1.07 µM, 4.19 µM, and 17.14 µM, respectively. CYP2C8 was inhibited by montelukast, montelukast sulfoxide, montelukast sulfone, tribendazole, triclabendazole sulfoxide, and triclabendazole sulfone with IC50 of 0.08 µM, 0.05 µM, 0.02 µM, 3.31 µM, 8.95 µM, and 1.05 µM, respectively. CYP2C9 was inhibited by triclabendazole, triclabendazole sulfoxide, triclabendazole sulfone, montelukast, montelukast sulfoxide and montelukast sulfone with IC50 of 1.17 µM, 1.95 µM, 0.69 µM, 1.34 µM, 3.61 µM and 2.15 µM, respectively. CYP2C19 was inhibited by triclabendazole and triclabendazole sulfoxide with IC50 of 0.25 and 0.22, respectively. CYP3A4 was inhibited by montelukast sulfoxide and triclabendazole with IC50 of 9.33 and 15.11, respectively. Amongst the studied sulfoxide/sulfone substrates, the propensity of involvement of CY2C9 and CYP3A4 enzyme was high (approximately 56% of total) in the metabolic fate experiments.
Conclusion: Based on the findings, a proper risk assessment strategy needs to be factored (i.e., perpetrator and/or victim drug) to overcome any imminent risk of potential clinical drug-drug interaction when sulfoxide/sulfone metabolite(s) generating drugs are coadministered in therapy.
期刊介绍:
Drug Metabolism Letters publishes letters and research articles on major advances in all areas of drug metabolism and disposition. The emphasis is on publishing quality papers very rapidly by taking full advantage of the Internet technology both for the submission and review of manuscripts. The journal covers the following areas: In vitro systems including CYP-450; enzyme induction and inhibition; drug-drug interactions and enzyme kinetics; pharmacokinetics, toxicokinetics, species scaling and extrapolations; P-glycoprotein and transport carriers; target organ toxicity and interindividual variability; drug metabolism and disposition studies; extrahepatic metabolism; phase I and phase II metabolism; recent developments for the identification of drug metabolites.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
