Machteld M Oud, Brooke L Latour, Zeineb Bakey, Stef J Letteboer, Dorien Lugtenberg, Ka Man Wu, Elisabeth A M Cornelissen, Helger G Yntema, Miriam Schmidts, Ronald Roepman, Ernie M H F Bongers
{"title":"细胞纤毛表型显示了IFT140新变异的致病性,并证实了Mainzer-Saldino综合征的诊断。","authors":"Machteld M Oud, Brooke L Latour, Zeineb Bakey, Stef J Letteboer, Dorien Lugtenberg, Ka Man Wu, Elisabeth A M Cornelissen, Helger G Yntema, Miriam Schmidts, Ronald Roepman, Ernie M H F Bongers","doi":"10.1186/s13630-018-0055-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mainzer-Saldino syndrome (MZSDS) is a skeletal ciliopathy and part of the short-rib thoracic dysplasia (SRTD) group of ciliary disorders. The main characteristics of MZSDS are short limbs, mild narrow thorax, blindness, and renal failure. Thus far, variants in two genes are associated with MZSDS: <i>IFT140,</i> and <i>IFT172</i>. In this study, we describe a 1-year-old girl presenting with mild skeletal abnormalities, Leber congenital amaurosis, and bilateral hearing difficulties. For establishing an accurate diagnosis, we combined clinical, molecular, and functional analyses.</p><p><strong>Methods: </strong>We performed diagnostic whole-exome sequencing (WES) analysis to determine the genetic cause of the disease and analyzed two gene panels, containing all currently known genes in vision disorders, and in hearing impairment. Upon detection of the likely causative variants, ciliary phenotyping was performed in patient urine-derived renal epithelial cells (URECs) and rescue experiments were performed in CRISPR/Cas9-derived <i>Ift140</i> knock out cells to determine the pathogenicity of the detected variants in vitro. Cilium morphology, cilium length, and intraflagellar transport (IFT) were evaluated by immunocytochemistry.</p><p><strong>Results: </strong>Diagnostic WES revealed two novel compound heterozygous variants in <i>IFT140</i>, encoding IFT140. Thorough investigation of WES data did not reveal any variants in candidate genes associated with hearing impairment. Patient-derived URECs revealed an accumulation of IFT-B protein IFT88 at the ciliary tip in 41% of the cells indicative of impaired retrograde IFT, while this was absent in cilia from control URECs. Furthermore, transfection of CRISPR/Cas9-derived <i>Ift140</i> knock out cells with an IFT140 construct containing the patient mutation p.Tyr923Asp resulted in a significantly higher percentage of IFT88 tip accumulation than transfection with the wild-type IFT140 construct.</p><p><strong>Conclusions: </strong>By combining the clinical, genetic, and functional data from this study, we could conclude that the patient has SRTD9, also called Mainzer-Saldino syndrome, caused by variants in <i>IFT140</i>. We suggest the possibility that variants in <i>IFT140</i> may underlie hearing impairment. Moreover, we show that urine provides an excellent source to obtain patient-derived cells in a non-invasive manner to study the pathogenicity of variants detected by genetic testing.</p>","PeriodicalId":38134,"journal":{"name":"Cilia","volume":"7 ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2018-02-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13630-018-0055-2","citationCount":"20","resultStr":"{\"title\":\"Cellular ciliary phenotyping indicates pathogenicity of novel variants in <i>IFT140</i> and confirms a Mainzer-Saldino syndrome diagnosis.\",\"authors\":\"Machteld M Oud, Brooke L Latour, Zeineb Bakey, Stef J Letteboer, Dorien Lugtenberg, Ka Man Wu, Elisabeth A M Cornelissen, Helger G Yntema, Miriam Schmidts, Ronald Roepman, Ernie M H F Bongers\",\"doi\":\"10.1186/s13630-018-0055-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mainzer-Saldino syndrome (MZSDS) is a skeletal ciliopathy and part of the short-rib thoracic dysplasia (SRTD) group of ciliary disorders. The main characteristics of MZSDS are short limbs, mild narrow thorax, blindness, and renal failure. Thus far, variants in two genes are associated with MZSDS: <i>IFT140,</i> and <i>IFT172</i>. In this study, we describe a 1-year-old girl presenting with mild skeletal abnormalities, Leber congenital amaurosis, and bilateral hearing difficulties. For establishing an accurate diagnosis, we combined clinical, molecular, and functional analyses.</p><p><strong>Methods: </strong>We performed diagnostic whole-exome sequencing (WES) analysis to determine the genetic cause of the disease and analyzed two gene panels, containing all currently known genes in vision disorders, and in hearing impairment. Upon detection of the likely causative variants, ciliary phenotyping was performed in patient urine-derived renal epithelial cells (URECs) and rescue experiments were performed in CRISPR/Cas9-derived <i>Ift140</i> knock out cells to determine the pathogenicity of the detected variants in vitro. Cilium morphology, cilium length, and intraflagellar transport (IFT) were evaluated by immunocytochemistry.</p><p><strong>Results: </strong>Diagnostic WES revealed two novel compound heterozygous variants in <i>IFT140</i>, encoding IFT140. Thorough investigation of WES data did not reveal any variants in candidate genes associated with hearing impairment. Patient-derived URECs revealed an accumulation of IFT-B protein IFT88 at the ciliary tip in 41% of the cells indicative of impaired retrograde IFT, while this was absent in cilia from control URECs. Furthermore, transfection of CRISPR/Cas9-derived <i>Ift140</i> knock out cells with an IFT140 construct containing the patient mutation p.Tyr923Asp resulted in a significantly higher percentage of IFT88 tip accumulation than transfection with the wild-type IFT140 construct.</p><p><strong>Conclusions: </strong>By combining the clinical, genetic, and functional data from this study, we could conclude that the patient has SRTD9, also called Mainzer-Saldino syndrome, caused by variants in <i>IFT140</i>. We suggest the possibility that variants in <i>IFT140</i> may underlie hearing impairment. Moreover, we show that urine provides an excellent source to obtain patient-derived cells in a non-invasive manner to study the pathogenicity of variants detected by genetic testing.</p>\",\"PeriodicalId\":38134,\"journal\":{\"name\":\"Cilia\",\"volume\":\"7 \",\"pages\":\"1\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-02-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13630-018-0055-2\",\"citationCount\":\"20\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cilia\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13630-018-0055-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cilia","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13630-018-0055-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Cellular ciliary phenotyping indicates pathogenicity of novel variants in IFT140 and confirms a Mainzer-Saldino syndrome diagnosis.
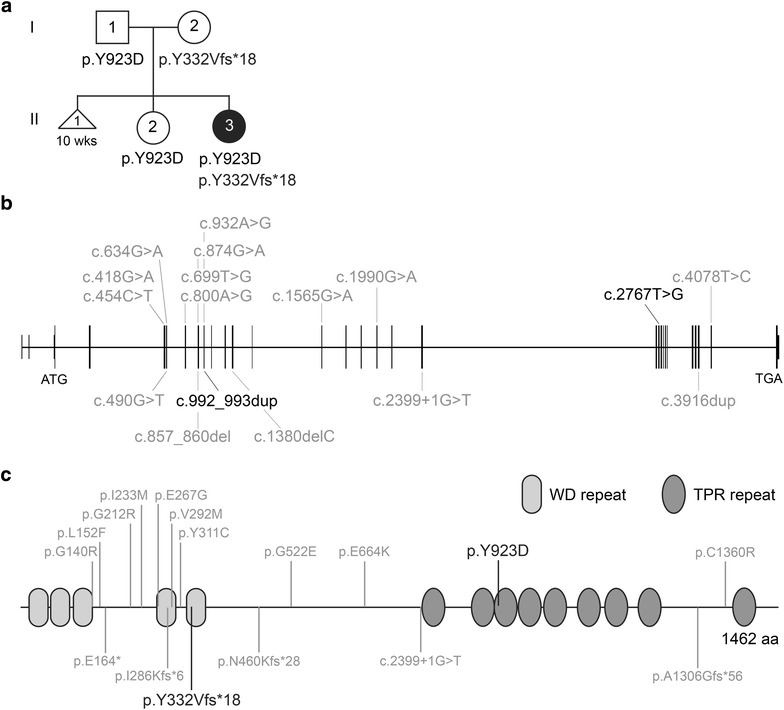
Background: Mainzer-Saldino syndrome (MZSDS) is a skeletal ciliopathy and part of the short-rib thoracic dysplasia (SRTD) group of ciliary disorders. The main characteristics of MZSDS are short limbs, mild narrow thorax, blindness, and renal failure. Thus far, variants in two genes are associated with MZSDS: IFT140, and IFT172. In this study, we describe a 1-year-old girl presenting with mild skeletal abnormalities, Leber congenital amaurosis, and bilateral hearing difficulties. For establishing an accurate diagnosis, we combined clinical, molecular, and functional analyses.
Methods: We performed diagnostic whole-exome sequencing (WES) analysis to determine the genetic cause of the disease and analyzed two gene panels, containing all currently known genes in vision disorders, and in hearing impairment. Upon detection of the likely causative variants, ciliary phenotyping was performed in patient urine-derived renal epithelial cells (URECs) and rescue experiments were performed in CRISPR/Cas9-derived Ift140 knock out cells to determine the pathogenicity of the detected variants in vitro. Cilium morphology, cilium length, and intraflagellar transport (IFT) were evaluated by immunocytochemistry.
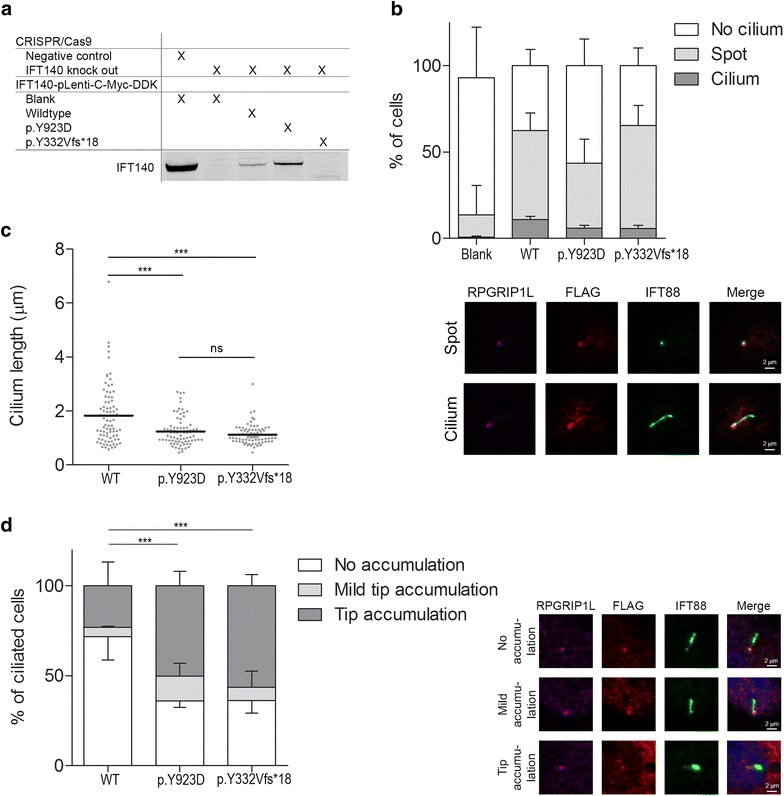
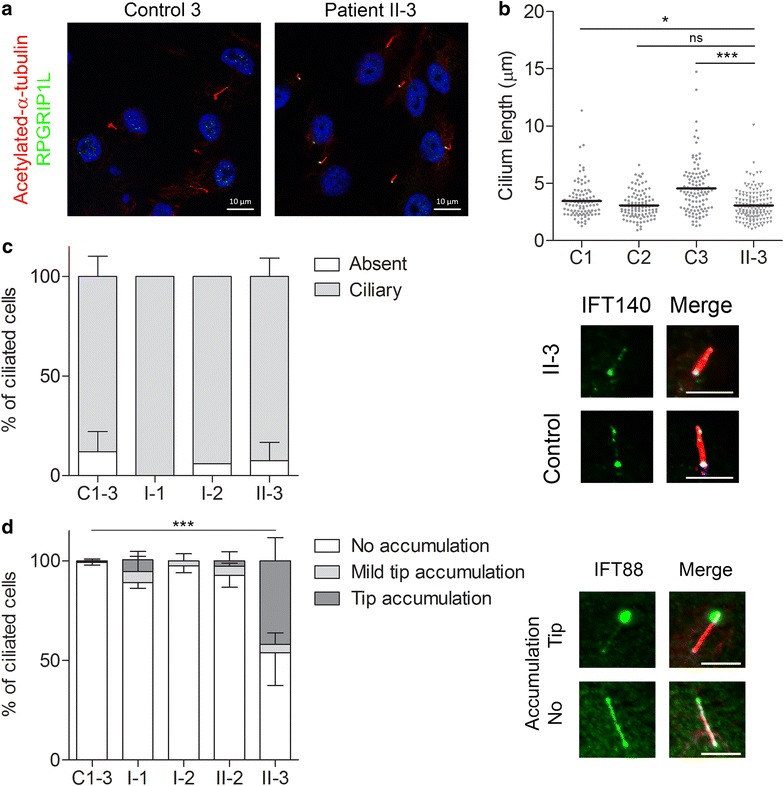
Results: Diagnostic WES revealed two novel compound heterozygous variants in IFT140, encoding IFT140. Thorough investigation of WES data did not reveal any variants in candidate genes associated with hearing impairment. Patient-derived URECs revealed an accumulation of IFT-B protein IFT88 at the ciliary tip in 41% of the cells indicative of impaired retrograde IFT, while this was absent in cilia from control URECs. Furthermore, transfection of CRISPR/Cas9-derived Ift140 knock out cells with an IFT140 construct containing the patient mutation p.Tyr923Asp resulted in a significantly higher percentage of IFT88 tip accumulation than transfection with the wild-type IFT140 construct.
Conclusions: By combining the clinical, genetic, and functional data from this study, we could conclude that the patient has SRTD9, also called Mainzer-Saldino syndrome, caused by variants in IFT140. We suggest the possibility that variants in IFT140 may underlie hearing impairment. Moreover, we show that urine provides an excellent source to obtain patient-derived cells in a non-invasive manner to study the pathogenicity of variants detected by genetic testing.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
