{"title":"研究孤儿疾病的人类iPSC模型:肌肉萎缩症。","authors":"Guangbin Xia, Naohiro Terada, Tetsuo Ashizawa","doi":"10.1007/s40778-018-0145-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose of review: </strong>Muscular dystrophies (MDs) are a spectrum of muscle disorders, which are caused by a number of gene mutations. The studies of MDs are limited due to lack of appropriate models, except for Duchenne muscular dystrophy (DMD), myotonic dystrophy type 1 (DM1), facioscapulohumeral muscular dystrophy (FSHD), and certain type of limb-girdle muscular dystrophy (LGMD). Human induced pluripotent stem cell (iPSC) technologies are emerging to offer a useful model for mechanistic studies, drug discovery, and cell-based therapy to supplement in vivo animal models. This review will focus on current applications of iPSC as disease models of MDs for studies of pathogenic mechanisms and therapeutic development.</p><p><strong>Recent findings: </strong>Many and more human disease-specific iPSCs have been or being established, which carry the natural mutation of MDs with human genomic background. These iPSCs can be differentiated into specific cell types affected in a particular MDs such as skeletal muscle progenitor cells, skeletal muscle fibers, and cardiomyocytes. Human iPSCs are particularly useful for studies of the pathogenicity at the early stage or developmental phase of MDs. High-throughput screening using disease-specific human iPSCs has become a powerful technology in drug discovery. While MD iPSCs have been generated for cell-based replacement therapy, recent advances in genome editing technologies enabled correction of genetic mutations in these cells in culture, raising hope for in vivo genome therapy, which offers a fundamental cure for these daunting inherited MDs.</p><p><strong>Summary: </strong>Human disease-specific iPSC models for MDs are emerging as an additional tool to current disease models for elucidating disease mechanisms and developing therapeutic intervention.</p>","PeriodicalId":37444,"journal":{"name":"Current Stem Cell Reports","volume":"4 4","pages":"299-309"},"PeriodicalIF":2.3000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1007/s40778-018-0145-5","citationCount":"13","resultStr":"{\"title\":\"Human iPSC Models to Study Orphan Diseases: Muscular Dystrophies.\",\"authors\":\"Guangbin Xia, Naohiro Terada, Tetsuo Ashizawa\",\"doi\":\"10.1007/s40778-018-0145-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose of review: </strong>Muscular dystrophies (MDs) are a spectrum of muscle disorders, which are caused by a number of gene mutations. The studies of MDs are limited due to lack of appropriate models, except for Duchenne muscular dystrophy (DMD), myotonic dystrophy type 1 (DM1), facioscapulohumeral muscular dystrophy (FSHD), and certain type of limb-girdle muscular dystrophy (LGMD). Human induced pluripotent stem cell (iPSC) technologies are emerging to offer a useful model for mechanistic studies, drug discovery, and cell-based therapy to supplement in vivo animal models. This review will focus on current applications of iPSC as disease models of MDs for studies of pathogenic mechanisms and therapeutic development.</p><p><strong>Recent findings: </strong>Many and more human disease-specific iPSCs have been or being established, which carry the natural mutation of MDs with human genomic background. These iPSCs can be differentiated into specific cell types affected in a particular MDs such as skeletal muscle progenitor cells, skeletal muscle fibers, and cardiomyocytes. Human iPSCs are particularly useful for studies of the pathogenicity at the early stage or developmental phase of MDs. High-throughput screening using disease-specific human iPSCs has become a powerful technology in drug discovery. While MD iPSCs have been generated for cell-based replacement therapy, recent advances in genome editing technologies enabled correction of genetic mutations in these cells in culture, raising hope for in vivo genome therapy, which offers a fundamental cure for these daunting inherited MDs.</p><p><strong>Summary: </strong>Human disease-specific iPSC models for MDs are emerging as an additional tool to current disease models for elucidating disease mechanisms and developing therapeutic intervention.</p>\",\"PeriodicalId\":37444,\"journal\":{\"name\":\"Current Stem Cell Reports\",\"volume\":\"4 4\",\"pages\":\"299-309\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2018-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1007/s40778-018-0145-5\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Stem Cell Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s40778-018-0145-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2018/10/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"CELL & TISSUE ENGINEERING\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Stem Cell Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s40778-018-0145-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/10/4 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CELL & TISSUE ENGINEERING","Score":null,"Total":0}
Human iPSC Models to Study Orphan Diseases: Muscular Dystrophies.
Purpose of review: Muscular dystrophies (MDs) are a spectrum of muscle disorders, which are caused by a number of gene mutations. The studies of MDs are limited due to lack of appropriate models, except for Duchenne muscular dystrophy (DMD), myotonic dystrophy type 1 (DM1), facioscapulohumeral muscular dystrophy (FSHD), and certain type of limb-girdle muscular dystrophy (LGMD). Human induced pluripotent stem cell (iPSC) technologies are emerging to offer a useful model for mechanistic studies, drug discovery, and cell-based therapy to supplement in vivo animal models. This review will focus on current applications of iPSC as disease models of MDs for studies of pathogenic mechanisms and therapeutic development.
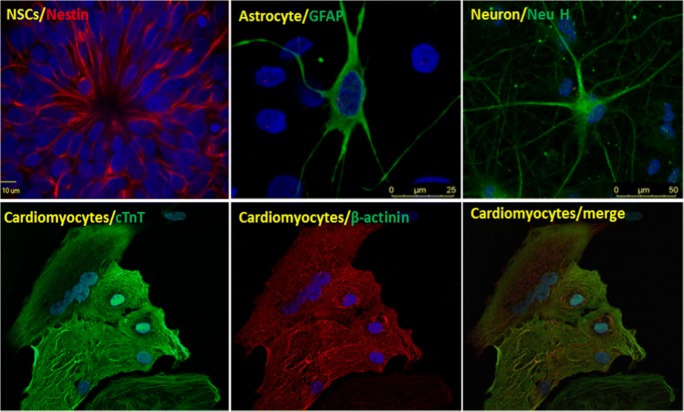
Recent findings: Many and more human disease-specific iPSCs have been or being established, which carry the natural mutation of MDs with human genomic background. These iPSCs can be differentiated into specific cell types affected in a particular MDs such as skeletal muscle progenitor cells, skeletal muscle fibers, and cardiomyocytes. Human iPSCs are particularly useful for studies of the pathogenicity at the early stage or developmental phase of MDs. High-throughput screening using disease-specific human iPSCs has become a powerful technology in drug discovery. While MD iPSCs have been generated for cell-based replacement therapy, recent advances in genome editing technologies enabled correction of genetic mutations in these cells in culture, raising hope for in vivo genome therapy, which offers a fundamental cure for these daunting inherited MDs.
Summary: Human disease-specific iPSC models for MDs are emerging as an additional tool to current disease models for elucidating disease mechanisms and developing therapeutic intervention.
期刊介绍:
The goal of this journal is to publish cutting-edge reviews on subjects pertinent to all aspects of stem cell research, therapy, ethics, commercialization, and policy. We aim to provide incisive, insightful, and balanced contributions from leading experts in each relevant domain that will be of immediate interest to a wide readership of clinicians, basic scientists, and translational investigators.
We accomplish this aim by appointing major authorities to serve as Section Editors in key subject areas across the discipline. Section Editors select topics to be reviewed by leading experts who emphasize recent developments and highlight important papers published over the past year on their topics, in a crisp and readable format. We also provide commentaries from well-known figures in the field, and an Editorial Board of internationally diverse members suggests topics of special interest to their country/region and ensures that topics are current and include emerging research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
