Yanyuan Dai, Yue Lang, Mingxuan Ding, Baizhuo Zhang, Xiaoou Han, Guangyu Duan, Li Cui
{"title":"罕见遗传性克雅氏病伴E196A突变1例","authors":"Yanyuan Dai, Yue Lang, Mingxuan Ding, Baizhuo Zhang, Xiaoou Han, Guangyu Duan, Li Cui","doi":"10.1080/19336896.2019.1631679","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic Creutzfeldt-Jakob disease (gCJD) accounts for approximately 10-15% of human prion diseases. It is an autosomal dominant disease caused by missense or insertion mutations of the gene that encodes prion protein (PRNP). In general, the manifestations and neuropathological changes of gCJD are similar to those of sporadic CJD (sCJD), and the diagnostic sensitivities of cerebrospinal fluid (CSF) markers, electroencephalography (EEG), and magnetic resonance imaging (MRI) are generally lower in gCJD than sCJD. Here we report on a 56-year-old Chinese woman who was diagnosed with gCJD and suspected to have thyroid cancer. The patient carried the glutamate to alanine substitution at codon 196 (E196A) of PRNP, which is quite a rare mutation and has only been reported in China. To our knowledge, this is the fourth case of E196A gCJD in the world. Here, we compared the manifestations and assistant examinations of the current patient with those of three previously reported Chinese patients with E196A gCJD in order to illustrate the common features of E196A gCJD.</p>","PeriodicalId":54585,"journal":{"name":"Prion","volume":"13 1","pages":"132-136"},"PeriodicalIF":1.6000,"publicationDate":"2019-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336896.2019.1631679","citationCount":"5","resultStr":"{\"title\":\"Rare genetic Creutzfeldt-Jakob disease with E196A mutation: a case report.\",\"authors\":\"Yanyuan Dai, Yue Lang, Mingxuan Ding, Baizhuo Zhang, Xiaoou Han, Guangyu Duan, Li Cui\",\"doi\":\"10.1080/19336896.2019.1631679\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Genetic Creutzfeldt-Jakob disease (gCJD) accounts for approximately 10-15% of human prion diseases. It is an autosomal dominant disease caused by missense or insertion mutations of the gene that encodes prion protein (PRNP). In general, the manifestations and neuropathological changes of gCJD are similar to those of sporadic CJD (sCJD), and the diagnostic sensitivities of cerebrospinal fluid (CSF) markers, electroencephalography (EEG), and magnetic resonance imaging (MRI) are generally lower in gCJD than sCJD. Here we report on a 56-year-old Chinese woman who was diagnosed with gCJD and suspected to have thyroid cancer. The patient carried the glutamate to alanine substitution at codon 196 (E196A) of PRNP, which is quite a rare mutation and has only been reported in China. To our knowledge, this is the fourth case of E196A gCJD in the world. Here, we compared the manifestations and assistant examinations of the current patient with those of three previously reported Chinese patients with E196A gCJD in order to illustrate the common features of E196A gCJD.</p>\",\"PeriodicalId\":54585,\"journal\":{\"name\":\"Prion\",\"volume\":\"13 1\",\"pages\":\"132-136\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2019-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1080/19336896.2019.1631679\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Prion\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1080/19336896.2019.1631679\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Prion","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1080/19336896.2019.1631679","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Rare genetic Creutzfeldt-Jakob disease with E196A mutation: a case report.
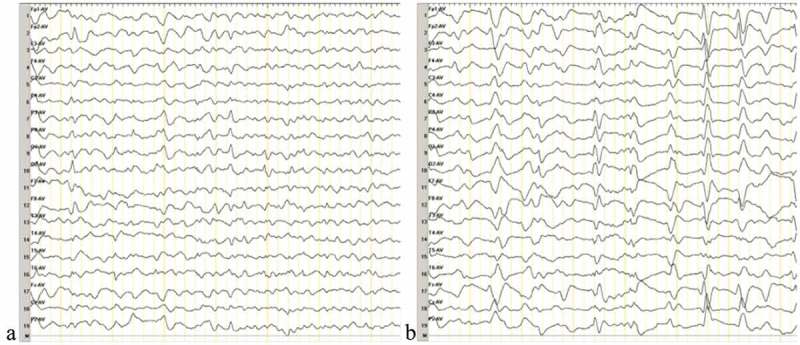
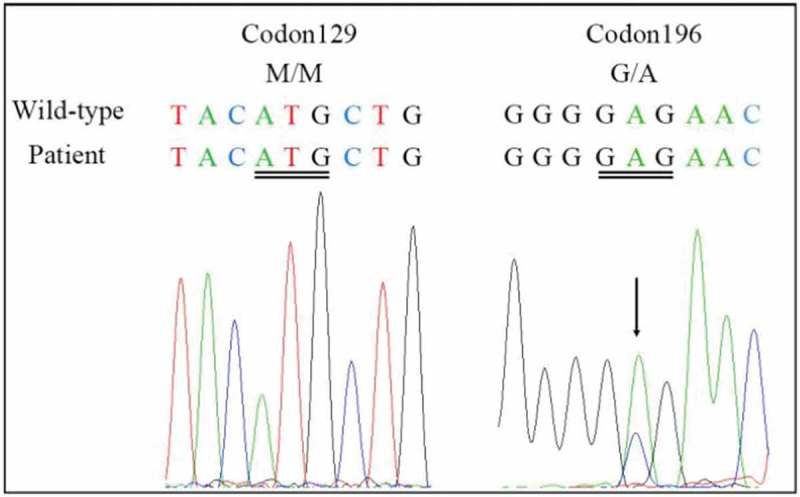
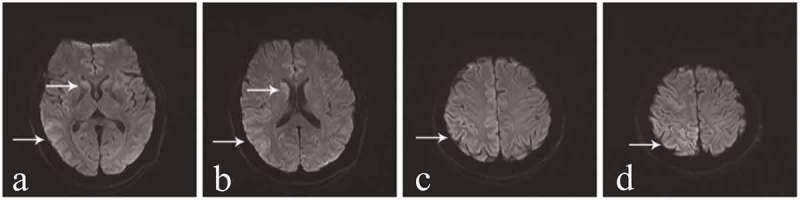
Genetic Creutzfeldt-Jakob disease (gCJD) accounts for approximately 10-15% of human prion diseases. It is an autosomal dominant disease caused by missense or insertion mutations of the gene that encodes prion protein (PRNP). In general, the manifestations and neuropathological changes of gCJD are similar to those of sporadic CJD (sCJD), and the diagnostic sensitivities of cerebrospinal fluid (CSF) markers, electroencephalography (EEG), and magnetic resonance imaging (MRI) are generally lower in gCJD than sCJD. Here we report on a 56-year-old Chinese woman who was diagnosed with gCJD and suspected to have thyroid cancer. The patient carried the glutamate to alanine substitution at codon 196 (E196A) of PRNP, which is quite a rare mutation and has only been reported in China. To our knowledge, this is the fourth case of E196A gCJD in the world. Here, we compared the manifestations and assistant examinations of the current patient with those of three previously reported Chinese patients with E196A gCJD in order to illustrate the common features of E196A gCJD.
期刊介绍:
Prion is the first international peer-reviewed open access journal to focus exclusively on protein folding and misfolding, protein assembly disorders, protein-based and structural inheritance. The goal is to foster communication and rapid exchange of information through timely publication of important results using traditional as well as electronic formats. The overriding criteria for publication in Prion are originality, scientific merit and general interest.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
