Georg Strebinger, Elena Müller, Alexandra Feldman, Elmar Aigner
{"title":"溶酶体酸性脂肪酶缺乏症-早期诊断是关键。","authors":"Georg Strebinger, Elena Müller, Alexandra Feldman, Elmar Aigner","doi":"10.2147/HMER.S201630","DOIUrl":null,"url":null,"abstract":"<p><p>Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease that may present from infancy to late adulthood depending on residual enzyme activity. While the severe form manifests as a rapidly progressive disease with near universal mortality within the first 6 months of life, milder forms frequently go undiagnosed for prolonged periods and typically present with progressive fatty liver disease, enlarged spleen, atherogenic dyslipidemia and premature atherosclerosis. The adult variant of LAL-D is typically diagnosed late or even overlooked due to the unspecific nature of the presenting symptoms, which are similar to common changes observed in the context of the metabolic syndrome. This review is aimed at delineating clinically useful scenarios in which pediatric or adult medicine clinicians should be aware of LAL-D as a differential diagnosis for selected patients. This is particularly relevant as a potentially life-saving enzyme replacement therapy has become available and the diagnosis can easily be ruled out or confirmed using a dried blood spot test.</p>","PeriodicalId":12917,"journal":{"name":"Hepatic Medicine : Evidence and Research","volume":"11 ","pages":"79-88"},"PeriodicalIF":1.8000,"publicationDate":"2019-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/HMER.S201630","citationCount":"23","resultStr":"{\"title\":\"Lysosomal acid lipase deficiency - early diagnosis is the key.\",\"authors\":\"Georg Strebinger, Elena Müller, Alexandra Feldman, Elmar Aigner\",\"doi\":\"10.2147/HMER.S201630\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease that may present from infancy to late adulthood depending on residual enzyme activity. While the severe form manifests as a rapidly progressive disease with near universal mortality within the first 6 months of life, milder forms frequently go undiagnosed for prolonged periods and typically present with progressive fatty liver disease, enlarged spleen, atherogenic dyslipidemia and premature atherosclerosis. The adult variant of LAL-D is typically diagnosed late or even overlooked due to the unspecific nature of the presenting symptoms, which are similar to common changes observed in the context of the metabolic syndrome. This review is aimed at delineating clinically useful scenarios in which pediatric or adult medicine clinicians should be aware of LAL-D as a differential diagnosis for selected patients. This is particularly relevant as a potentially life-saving enzyme replacement therapy has become available and the diagnosis can easily be ruled out or confirmed using a dried blood spot test.</p>\",\"PeriodicalId\":12917,\"journal\":{\"name\":\"Hepatic Medicine : Evidence and Research\",\"volume\":\"11 \",\"pages\":\"79-88\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2019-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2147/HMER.S201630\",\"citationCount\":\"23\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hepatic Medicine : Evidence and Research\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/HMER.S201630\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GASTROENTEROLOGY & HEPATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hepatic Medicine : Evidence and Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/HMER.S201630","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
Lysosomal acid lipase deficiency - early diagnosis is the key.
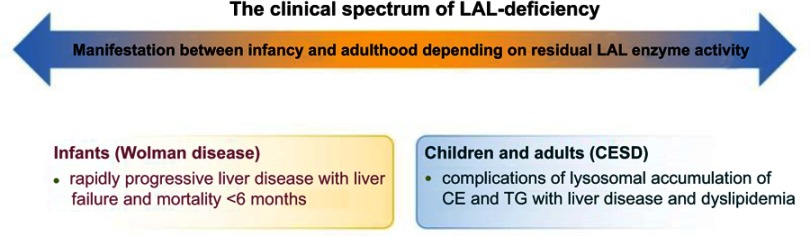
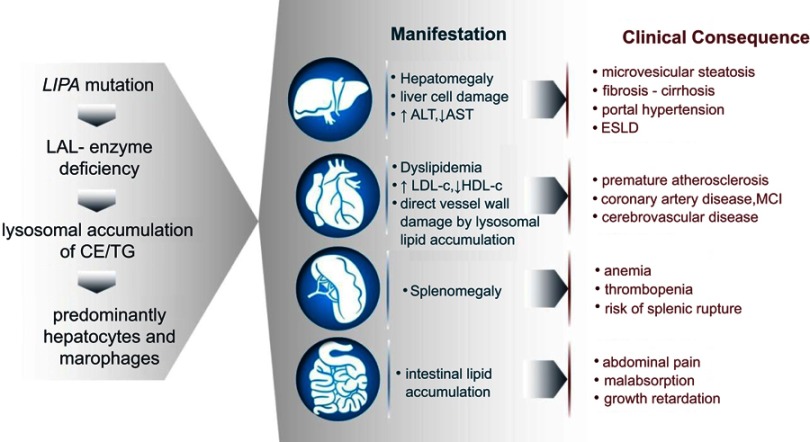
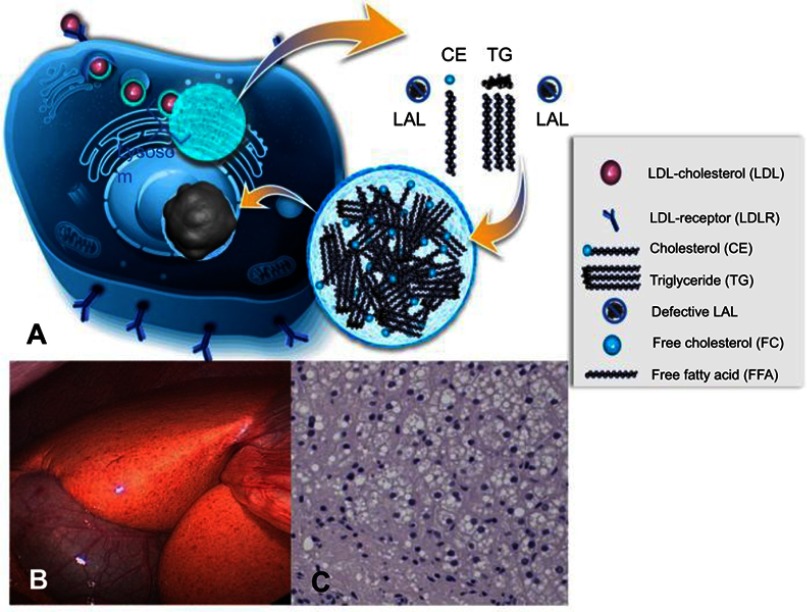
Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease that may present from infancy to late adulthood depending on residual enzyme activity. While the severe form manifests as a rapidly progressive disease with near universal mortality within the first 6 months of life, milder forms frequently go undiagnosed for prolonged periods and typically present with progressive fatty liver disease, enlarged spleen, atherogenic dyslipidemia and premature atherosclerosis. The adult variant of LAL-D is typically diagnosed late or even overlooked due to the unspecific nature of the presenting symptoms, which are similar to common changes observed in the context of the metabolic syndrome. This review is aimed at delineating clinically useful scenarios in which pediatric or adult medicine clinicians should be aware of LAL-D as a differential diagnosis for selected patients. This is particularly relevant as a potentially life-saving enzyme replacement therapy has become available and the diagnosis can easily be ruled out or confirmed using a dried blood spot test.
期刊介绍:
Hepatic Medicine: Evidence and Research is an international, peer-reviewed, open access, online journal. Publishing original research, reports, editorials, reviews and commentaries on all aspects of adult and pediatric hepatology in the clinic and laboratory including the following topics: Pathology, pathophysiology of hepatic disease Investigation and treatment of hepatic disease Pharmacology of drugs used for the treatment of hepatic disease Although the main focus of the journal is to publish research and clinical results in humans; preclinical, animal and in vitro studies will be published where they will shed light on disease processes and potential new therapies. Issues of patient safety and quality of care will also be considered. As of 1st April 2019, Hepatic Medicine: Evidence and Research will no longer consider meta-analyses for publication.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
