Lucy H Maynard, Olivia Smith, Nicolas P Tilmans, Eleonore Tham, Shayan Hosseinzadeh, Weilun Tan, Ryan Leenay, Andrew P May, Nicole K Paulk
{"title":"Fast-Seq:一种在学术环境中快速廉价验证包装单链腺相关病毒基因组的简单方法。","authors":"Lucy H Maynard, Olivia Smith, Nicolas P Tilmans, Eleonore Tham, Shayan Hosseinzadeh, Weilun Tan, Ryan Leenay, Andrew P May, Nicole K Paulk","doi":"10.1089/hgtb.2019.110","DOIUrl":null,"url":null,"abstract":"<p><p>Adeno-associated viral (AAV) vectors have shown great promise in gene delivery as evidenced by recent FDA approvals. Despite efforts to optimize manufacturing for good manufacturing practice (GMP) productions, few academic laboratories have the resources to assess vector composition. One critical component of vector quality is packaged genome fidelity. Errors in viral genome replication and packaging can result in the incorporation of faulty genomes with mutations, truncations, or rearrangements, compromising vector potency. Thus, sequence validation of packaged genome composition is an important quality control (QC), even in academic settings. We developed Fast-Seq, an end-to-end method for extraction, purification, sequencing, and data analysis of packaged single-stranded AAV (ssAAV) genomes intended for non-GMP preclinical environments. We validated Fast-Seq on ssAAV vectors with three different genome compositions (CAG-GFP, CAG-tdTomato, EF1α-FLuc), three different genome sizes (2.9, 3.6, 4.4 kb), packaged in four different capsid serotypes (AAV1, AAV2, AAV5, and AAV8), and produced using the two most common production methods (Baculovirus-<i>Sf9</i> and human HEK293), from both common commercial vendors and academic core facilities supplying academic laboratories. We achieved an average genome coverage of >1,400 × and an average inverted terminal repeat coverage of >280 × , despite the many differences in composition of each ssAAV sample. When compared with other ssAAV next-generation sequencing (NGS) methods for GMP settings, Fast-Seq has several unique advantages: Tn5 transposase-based fragmentation rather than sonication, 125 × less input DNA, simpler adapter ligation, compatibility with commonly available inexpensive sequencing instruments, and free open-source data analysis code in a preassembled customizable Docker container designed for novices. Fast-Seq can be completed in 18 h, is more cost-effective than other NGS methods, and is more accurate than Sanger sequencing, which is generally only applied at 1-2 × sequencing depth. Fast-Seq is a rapid, simple, and inexpensive methodology to validate packaged ssAAV genomes in academic settings.</p>","PeriodicalId":13126,"journal":{"name":"Human Gene Therapy Methods","volume":" ","pages":"195-205"},"PeriodicalIF":0.0000,"publicationDate":"2019-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1089/hgtb.2019.110","citationCount":"10","resultStr":"{\"title\":\"Fast-Seq: A Simple Method for Rapid and Inexpensive Validation of Packaged Single-Stranded Adeno-Associated Viral Genomes in Academic Settings.\",\"authors\":\"Lucy H Maynard, Olivia Smith, Nicolas P Tilmans, Eleonore Tham, Shayan Hosseinzadeh, Weilun Tan, Ryan Leenay, Andrew P May, Nicole K Paulk\",\"doi\":\"10.1089/hgtb.2019.110\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Adeno-associated viral (AAV) vectors have shown great promise in gene delivery as evidenced by recent FDA approvals. Despite efforts to optimize manufacturing for good manufacturing practice (GMP) productions, few academic laboratories have the resources to assess vector composition. One critical component of vector quality is packaged genome fidelity. Errors in viral genome replication and packaging can result in the incorporation of faulty genomes with mutations, truncations, or rearrangements, compromising vector potency. Thus, sequence validation of packaged genome composition is an important quality control (QC), even in academic settings. We developed Fast-Seq, an end-to-end method for extraction, purification, sequencing, and data analysis of packaged single-stranded AAV (ssAAV) genomes intended for non-GMP preclinical environments. We validated Fast-Seq on ssAAV vectors with three different genome compositions (CAG-GFP, CAG-tdTomato, EF1α-FLuc), three different genome sizes (2.9, 3.6, 4.4 kb), packaged in four different capsid serotypes (AAV1, AAV2, AAV5, and AAV8), and produced using the two most common production methods (Baculovirus-<i>Sf9</i> and human HEK293), from both common commercial vendors and academic core facilities supplying academic laboratories. We achieved an average genome coverage of >1,400 × and an average inverted terminal repeat coverage of >280 × , despite the many differences in composition of each ssAAV sample. When compared with other ssAAV next-generation sequencing (NGS) methods for GMP settings, Fast-Seq has several unique advantages: Tn5 transposase-based fragmentation rather than sonication, 125 × less input DNA, simpler adapter ligation, compatibility with commonly available inexpensive sequencing instruments, and free open-source data analysis code in a preassembled customizable Docker container designed for novices. Fast-Seq can be completed in 18 h, is more cost-effective than other NGS methods, and is more accurate than Sanger sequencing, which is generally only applied at 1-2 × sequencing depth. Fast-Seq is a rapid, simple, and inexpensive methodology to validate packaged ssAAV genomes in academic settings.</p>\",\"PeriodicalId\":13126,\"journal\":{\"name\":\"Human Gene Therapy Methods\",\"volume\":\" \",\"pages\":\"195-205\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2019-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1089/hgtb.2019.110\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Gene Therapy Methods\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1089/hgtb.2019.110\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Immunology and Microbiology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Gene Therapy Methods","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1089/hgtb.2019.110","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Immunology and Microbiology","Score":null,"Total":0}
Fast-Seq: A Simple Method for Rapid and Inexpensive Validation of Packaged Single-Stranded Adeno-Associated Viral Genomes in Academic Settings.
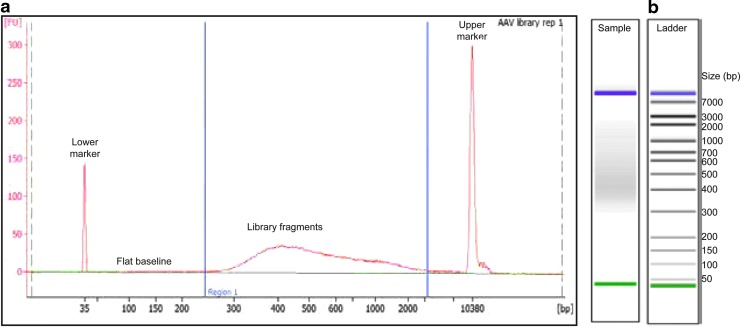
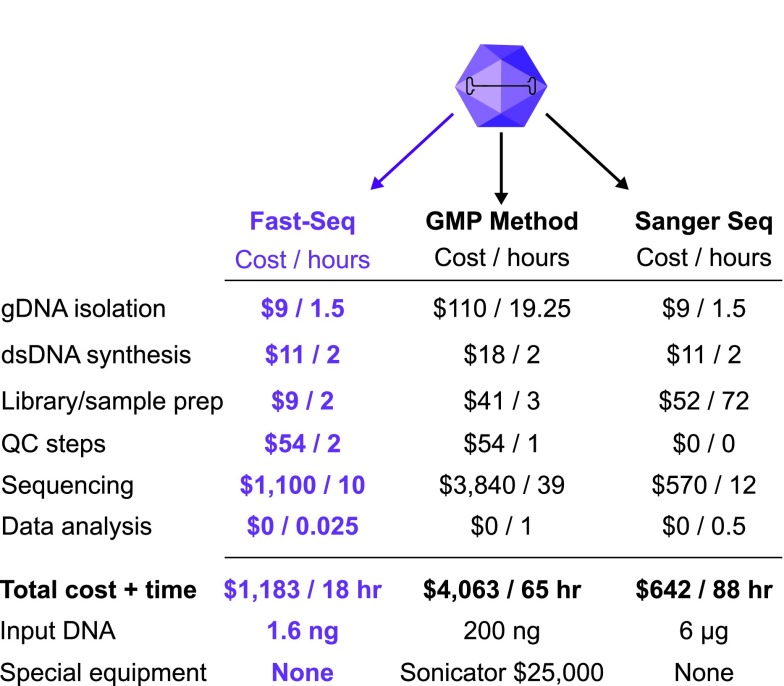
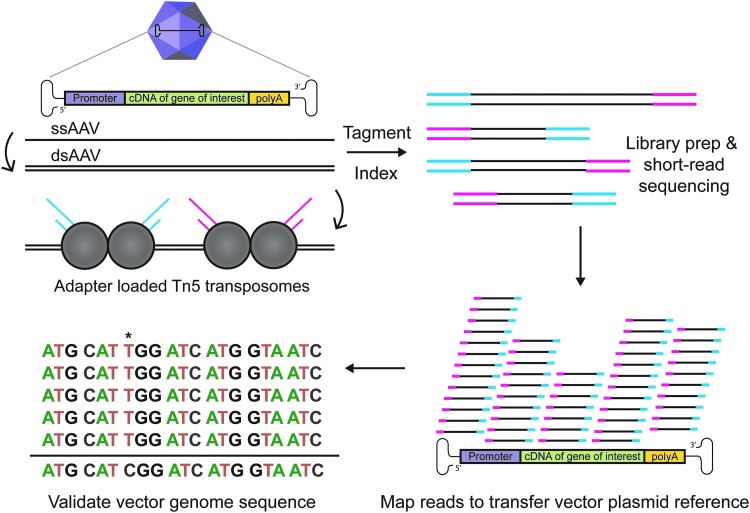
Adeno-associated viral (AAV) vectors have shown great promise in gene delivery as evidenced by recent FDA approvals. Despite efforts to optimize manufacturing for good manufacturing practice (GMP) productions, few academic laboratories have the resources to assess vector composition. One critical component of vector quality is packaged genome fidelity. Errors in viral genome replication and packaging can result in the incorporation of faulty genomes with mutations, truncations, or rearrangements, compromising vector potency. Thus, sequence validation of packaged genome composition is an important quality control (QC), even in academic settings. We developed Fast-Seq, an end-to-end method for extraction, purification, sequencing, and data analysis of packaged single-stranded AAV (ssAAV) genomes intended for non-GMP preclinical environments. We validated Fast-Seq on ssAAV vectors with three different genome compositions (CAG-GFP, CAG-tdTomato, EF1α-FLuc), three different genome sizes (2.9, 3.6, 4.4 kb), packaged in four different capsid serotypes (AAV1, AAV2, AAV5, and AAV8), and produced using the two most common production methods (Baculovirus-Sf9 and human HEK293), from both common commercial vendors and academic core facilities supplying academic laboratories. We achieved an average genome coverage of >1,400 × and an average inverted terminal repeat coverage of >280 × , despite the many differences in composition of each ssAAV sample. When compared with other ssAAV next-generation sequencing (NGS) methods for GMP settings, Fast-Seq has several unique advantages: Tn5 transposase-based fragmentation rather than sonication, 125 × less input DNA, simpler adapter ligation, compatibility with commonly available inexpensive sequencing instruments, and free open-source data analysis code in a preassembled customizable Docker container designed for novices. Fast-Seq can be completed in 18 h, is more cost-effective than other NGS methods, and is more accurate than Sanger sequencing, which is generally only applied at 1-2 × sequencing depth. Fast-Seq is a rapid, simple, and inexpensive methodology to validate packaged ssAAV genomes in academic settings.
期刊介绍:
Human Gene Therapy is the premier, multidisciplinary journal covering all aspects of gene therapy. The Journal publishes in-depth coverage of DNA, RNA, and cell therapies by delivering the latest breakthroughs in research and technologies. Human Gene Therapy provides a central forum for scientific and clinical information, including ethical, legal, regulatory, social, and commercial issues, which enables the advancement and progress of therapeutic procedures leading to improved patient outcomes, and ultimately, to curing diseases.
The Journal is divided into three parts. Human Gene Therapy, the flagship, is published 12 times per year. HGT Methods, a bimonthly journal, focuses on the applications of gene therapy to product testing and development. HGT Clinical Development, a quarterly journal, serves as a venue for publishing data relevant to the regulatory review and commercial development of cell and gene therapy products.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
