Ingrid Bader, Nina McTiernan, Christine Darbakk, Eugen Boltshauser, Rasmus Ree, Sabine Ebner, Johannes A Mayr, Thomas Arnesen
{"title":"一名患有 NAA10 功能障碍和新型杂合子新生 NAA10 p.(His16Pro) 变异的女孩的严重综合征 ID 和偏斜 X 失活 - 病例报告。","authors":"Ingrid Bader, Nina McTiernan, Christine Darbakk, Eugen Boltshauser, Rasmus Ree, Sabine Ebner, Johannes A Mayr, Thomas Arnesen","doi":"10.1186/s12881-020-01091-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>NAA10 is the catalytic subunit of the major N-terminal acetyltransferase complex NatA which acetylates almost half the human proteome. Over the past decade, many NAA10 missense variants have been reported as causative of genetic disease in humans. Individuals harboring NAA10 variants often display variable degrees of intellectual disability (ID), developmental delay, and cardiac anomalies. Initially, carrier females appeared to be oligo- or asymptomatic with X-inactivation pattern skewed towards the wild type allele. However, recently it has been shown that NAA10 variants can cause syndromic or non-syndromic intellectual disability in females as well. The impact of specific NAA10 variants and the X-inactivation pattern on the individual phenotype in females remains to be elucidated.</p><p><strong>Case presentation: </strong>Here we present a novel de novo NAA10 (NM_003491.3) c.[47A > C];[=] (p.[His16Pro];[=]) variant identified in a young female. The 10-year-old girl has severely delayed motor and language development, disturbed behavior with hyperactivity and restlessness, moderate dilatation of the ventricular system and extracerebral CSF spaces. Her blood leukocyte X-inactivation pattern was skewed (95/5) towards the maternally inherited X-chromosome. Our functional study indicates that NAA10 p.(H16P) impairs NatA complex formation and NatA catalytic activity, while monomeric NAA10 catalytic activity appears to be intact. Furthermore, cycloheximide experiments show that the NAA10 H16P variant does not affect the cellular stability of NAA10.</p><p><strong>Discussion and conclusions: </strong>We demonstrate that NAA10 p.(His16Pro) causes a severe form of syndromic ID in a girl most likely through impaired NatA-mediated Nt-acetylation of cellular proteins. X-inactivation analyses showed a skewed X-inactivation pattern in DNA from blood of the patient with the maternally inherited allele being preferentially methylated/inactivated.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"153"},"PeriodicalIF":0.0000,"publicationDate":"2020-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7374887/pdf/","citationCount":"0","resultStr":"{\"title\":\"Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant - a case report.\",\"authors\":\"Ingrid Bader, Nina McTiernan, Christine Darbakk, Eugen Boltshauser, Rasmus Ree, Sabine Ebner, Johannes A Mayr, Thomas Arnesen\",\"doi\":\"10.1186/s12881-020-01091-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>NAA10 is the catalytic subunit of the major N-terminal acetyltransferase complex NatA which acetylates almost half the human proteome. Over the past decade, many NAA10 missense variants have been reported as causative of genetic disease in humans. Individuals harboring NAA10 variants often display variable degrees of intellectual disability (ID), developmental delay, and cardiac anomalies. Initially, carrier females appeared to be oligo- or asymptomatic with X-inactivation pattern skewed towards the wild type allele. However, recently it has been shown that NAA10 variants can cause syndromic or non-syndromic intellectual disability in females as well. The impact of specific NAA10 variants and the X-inactivation pattern on the individual phenotype in females remains to be elucidated.</p><p><strong>Case presentation: </strong>Here we present a novel de novo NAA10 (NM_003491.3) c.[47A > C];[=] (p.[His16Pro];[=]) variant identified in a young female. The 10-year-old girl has severely delayed motor and language development, disturbed behavior with hyperactivity and restlessness, moderate dilatation of the ventricular system and extracerebral CSF spaces. Her blood leukocyte X-inactivation pattern was skewed (95/5) towards the maternally inherited X-chromosome. Our functional study indicates that NAA10 p.(H16P) impairs NatA complex formation and NatA catalytic activity, while monomeric NAA10 catalytic activity appears to be intact. Furthermore, cycloheximide experiments show that the NAA10 H16P variant does not affect the cellular stability of NAA10.</p><p><strong>Discussion and conclusions: </strong>We demonstrate that NAA10 p.(His16Pro) causes a severe form of syndromic ID in a girl most likely through impaired NatA-mediated Nt-acetylation of cellular proteins. X-inactivation analyses showed a skewed X-inactivation pattern in DNA from blood of the patient with the maternally inherited allele being preferentially methylated/inactivated.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"153\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-07-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7374887/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01091-1\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01091-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
摘要
背景:NAA10是主要N-端乙酰转移酶复合体NatA的催化亚基,NatA可使几乎一半的人类蛋白质组发生乙酰化。在过去的十年中,许多 NAA10 错义变体被报道可导致人类遗传疾病。携带NAA10变异体的个体通常表现出不同程度的智力障碍(ID)、发育迟缓和心脏异常。最初,女性携带者似乎没有症状或症状不明显,X-失活模式偏向野生型等位基因。然而,最近的研究表明,NAA10变体也可导致女性综合征或非综合征性智障。特定的 NAA10 变异和 X 失活模式对女性个体表型的影响仍有待阐明:在这里,我们介绍了在一名年轻女性身上发现的一个新的NAA10 (NM_003491.3) c.[47A > C];[=] (p.[His16Pro];[=]) 变异。这名 10 岁女孩的运动和语言发育严重迟缓,行为紊乱,多动不安,脑室系统和脑脊液外间隙中度扩张。她的血液白细胞 X-失活模式(95/5)偏向于母体遗传的 X 染色体。我们的功能研究表明,NAA10 p.(H16P) 会损害 NatA 复合物的形成和 NatA 的催化活性,而单体 NAA10 的催化活性似乎完好无损。此外,环己亚胺实验表明,NAA10 H16P变体不会影响NAA10的细胞稳定性:我们证明,NAA10 p.(His16Pro)最有可能是通过NatA介导的细胞蛋白Nt-乙酰化功能受损而导致一名女孩出现严重的综合症。X失活分析表明,患者血液中的DNA存在偏斜的X失活模式,母系遗传的等位基因优先被甲基化/失活。
Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant - a case report.
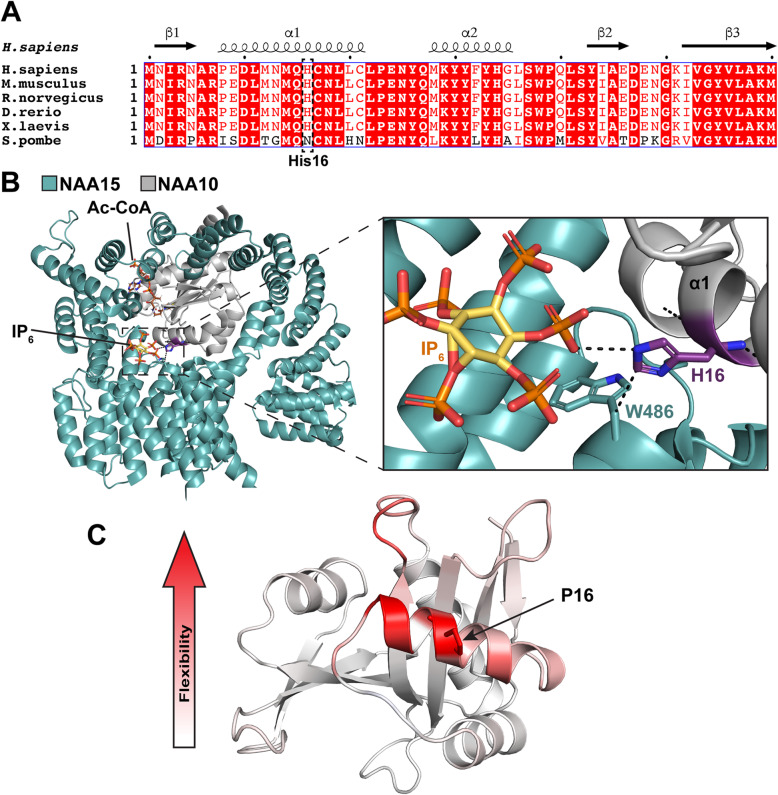
Background: NAA10 is the catalytic subunit of the major N-terminal acetyltransferase complex NatA which acetylates almost half the human proteome. Over the past decade, many NAA10 missense variants have been reported as causative of genetic disease in humans. Individuals harboring NAA10 variants often display variable degrees of intellectual disability (ID), developmental delay, and cardiac anomalies. Initially, carrier females appeared to be oligo- or asymptomatic with X-inactivation pattern skewed towards the wild type allele. However, recently it has been shown that NAA10 variants can cause syndromic or non-syndromic intellectual disability in females as well. The impact of specific NAA10 variants and the X-inactivation pattern on the individual phenotype in females remains to be elucidated.
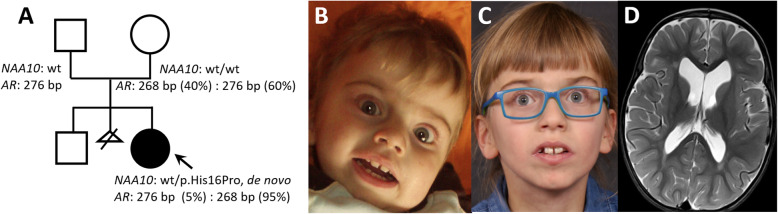
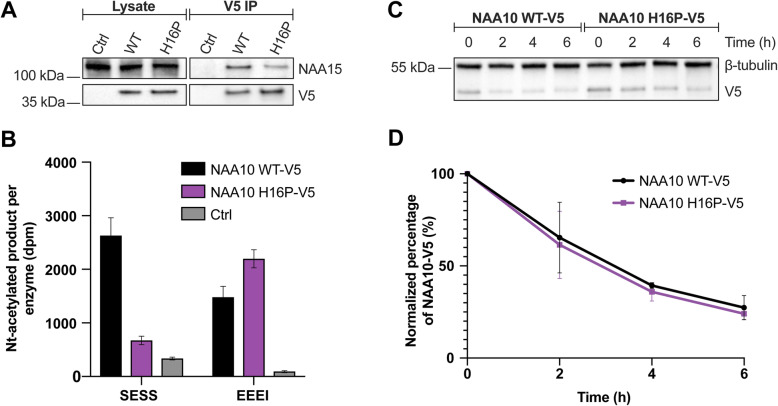
Case presentation: Here we present a novel de novo NAA10 (NM_003491.3) c.[47A > C];[=] (p.[His16Pro];[=]) variant identified in a young female. The 10-year-old girl has severely delayed motor and language development, disturbed behavior with hyperactivity and restlessness, moderate dilatation of the ventricular system and extracerebral CSF spaces. Her blood leukocyte X-inactivation pattern was skewed (95/5) towards the maternally inherited X-chromosome. Our functional study indicates that NAA10 p.(H16P) impairs NatA complex formation and NatA catalytic activity, while monomeric NAA10 catalytic activity appears to be intact. Furthermore, cycloheximide experiments show that the NAA10 H16P variant does not affect the cellular stability of NAA10.
Discussion and conclusions: We demonstrate that NAA10 p.(His16Pro) causes a severe form of syndromic ID in a girl most likely through impaired NatA-mediated Nt-acetylation of cellular proteins. X-inactivation analyses showed a skewed X-inactivation pattern in DNA from blood of the patient with the maternally inherited allele being preferentially methylated/inactivated.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
