{"title":"利用分类和回归方法从基因表达数据预测RNA甲基化状态。","authors":"Hao Xue, Zhen Wei, Kunqi Chen, Yujiao Tang, Xiangyu Wu, Jionglong Su, Jia Meng","doi":"10.1177/1176934320915707","DOIUrl":null,"url":null,"abstract":"<p><p>RNA <i>N</i> <sup>6</sup>-methyladenosine (m<sup>6</sup>A) has emerged as an important epigenetic modification for its role in regulating the stability, structure, processing, and translation of RNA. Instability of m<sup>6</sup>A homeostasis may result in flaws in stem cell regulation, decrease in fertility, and risk of cancer. To this day, experimental detection and quantification of RNA m<sup>6</sup>A modification are still time-consuming and labor-intensive. There is only a limited number of epitranscriptome samples in existing databases, and a matched RNA methylation profile is not often available for a biological problem of interests. As gene expression data are usually readily available for most biological problems, it could be appealing if we can estimate the RNA methylation status from gene expression data using <i>in silico</i> methods. In this study, we explored the possibility of computational prediction of RNA methylation status from gene expression data using classification and regression methods based on mouse RNA methylation data collected from 73 experimental conditions. Elastic Net-regularized Logistic Regression (ENLR), Support Vector Machine (SVM), and Random Forests (RF) were constructed for classification. Both SVM and RF achieved the best performance with the mean area under the curve (AUC) = 0.84 across samples; SVM had a narrower AUC spread. Gene Site Enrichment Analysis was conducted on those sites selected by ENLR as predictors to access the biological significance of the model. Three functional annotation terms were found statistically significant: phosphoprotein, SRC Homology 3 (SH3) domain, and endoplasmic reticulum. All 3 terms were found to be closely related to m<sup>6</sup>A pathway. For regression analysis, Elastic Net was implemented, which yielded a mean Pearson correlation coefficient = 0.68 and a mean Spearman correlation coefficient = 0.64. Our exploratory study suggested that gene expression data could be used to construct predictors for m<sup>6</sup>A methylation status with adequate accuracy. Our work showed for the first time that RNA methylation status may be predicted from the matched gene expression data. This finding may facilitate RNA modification research in various biological contexts when a matched RNA methylation profile is not available, especially in the very early stage of the study.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"16 ","pages":"1176934320915707"},"PeriodicalIF":1.6000,"publicationDate":"2020-07-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/1176934320915707","citationCount":"5","resultStr":"{\"title\":\"Prediction of RNA Methylation Status From Gene Expression Data Using Classification and Regression Methods.\",\"authors\":\"Hao Xue, Zhen Wei, Kunqi Chen, Yujiao Tang, Xiangyu Wu, Jionglong Su, Jia Meng\",\"doi\":\"10.1177/1176934320915707\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>RNA <i>N</i> <sup>6</sup>-methyladenosine (m<sup>6</sup>A) has emerged as an important epigenetic modification for its role in regulating the stability, structure, processing, and translation of RNA. Instability of m<sup>6</sup>A homeostasis may result in flaws in stem cell regulation, decrease in fertility, and risk of cancer. To this day, experimental detection and quantification of RNA m<sup>6</sup>A modification are still time-consuming and labor-intensive. There is only a limited number of epitranscriptome samples in existing databases, and a matched RNA methylation profile is not often available for a biological problem of interests. As gene expression data are usually readily available for most biological problems, it could be appealing if we can estimate the RNA methylation status from gene expression data using <i>in silico</i> methods. In this study, we explored the possibility of computational prediction of RNA methylation status from gene expression data using classification and regression methods based on mouse RNA methylation data collected from 73 experimental conditions. Elastic Net-regularized Logistic Regression (ENLR), Support Vector Machine (SVM), and Random Forests (RF) were constructed for classification. Both SVM and RF achieved the best performance with the mean area under the curve (AUC) = 0.84 across samples; SVM had a narrower AUC spread. Gene Site Enrichment Analysis was conducted on those sites selected by ENLR as predictors to access the biological significance of the model. Three functional annotation terms were found statistically significant: phosphoprotein, SRC Homology 3 (SH3) domain, and endoplasmic reticulum. All 3 terms were found to be closely related to m<sup>6</sup>A pathway. For regression analysis, Elastic Net was implemented, which yielded a mean Pearson correlation coefficient = 0.68 and a mean Spearman correlation coefficient = 0.64. Our exploratory study suggested that gene expression data could be used to construct predictors for m<sup>6</sup>A methylation status with adequate accuracy. Our work showed for the first time that RNA methylation status may be predicted from the matched gene expression data. This finding may facilitate RNA modification research in various biological contexts when a matched RNA methylation profile is not available, especially in the very early stage of the study.</p>\",\"PeriodicalId\":50472,\"journal\":{\"name\":\"Evolutionary Bioinformatics\",\"volume\":\"16 \",\"pages\":\"1176934320915707\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2020-07-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1177/1176934320915707\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Evolutionary Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1177/1176934320915707\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/1176934320915707","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
Prediction of RNA Methylation Status From Gene Expression Data Using Classification and Regression Methods.
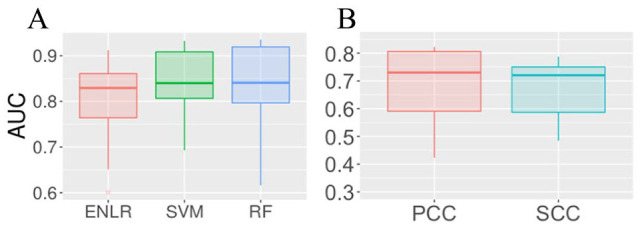
RNA N6-methyladenosine (m6A) has emerged as an important epigenetic modification for its role in regulating the stability, structure, processing, and translation of RNA. Instability of m6A homeostasis may result in flaws in stem cell regulation, decrease in fertility, and risk of cancer. To this day, experimental detection and quantification of RNA m6A modification are still time-consuming and labor-intensive. There is only a limited number of epitranscriptome samples in existing databases, and a matched RNA methylation profile is not often available for a biological problem of interests. As gene expression data are usually readily available for most biological problems, it could be appealing if we can estimate the RNA methylation status from gene expression data using in silico methods. In this study, we explored the possibility of computational prediction of RNA methylation status from gene expression data using classification and regression methods based on mouse RNA methylation data collected from 73 experimental conditions. Elastic Net-regularized Logistic Regression (ENLR), Support Vector Machine (SVM), and Random Forests (RF) were constructed for classification. Both SVM and RF achieved the best performance with the mean area under the curve (AUC) = 0.84 across samples; SVM had a narrower AUC spread. Gene Site Enrichment Analysis was conducted on those sites selected by ENLR as predictors to access the biological significance of the model. Three functional annotation terms were found statistically significant: phosphoprotein, SRC Homology 3 (SH3) domain, and endoplasmic reticulum. All 3 terms were found to be closely related to m6A pathway. For regression analysis, Elastic Net was implemented, which yielded a mean Pearson correlation coefficient = 0.68 and a mean Spearman correlation coefficient = 0.64. Our exploratory study suggested that gene expression data could be used to construct predictors for m6A methylation status with adequate accuracy. Our work showed for the first time that RNA methylation status may be predicted from the matched gene expression data. This finding may facilitate RNA modification research in various biological contexts when a matched RNA methylation profile is not available, especially in the very early stage of the study.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.
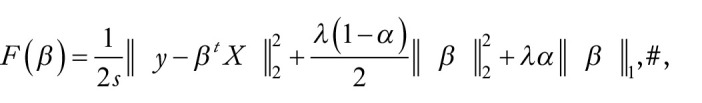
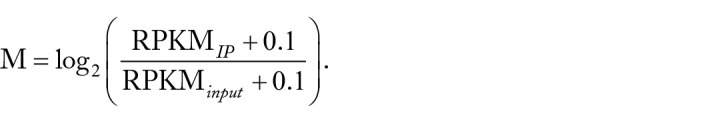




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
