D Hettiarachchi, Hetalkumar Panchal, P S Lai, V H W Dissanayake
{"title":"与儿童综合征和并指畸形相关的NSDHL基因新变异1例报告。","authors":"D Hettiarachchi, Hetalkumar Panchal, P S Lai, V H W Dissanayake","doi":"10.1186/s12881-020-01094-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.</p><p><strong>Case presentation: </strong>The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).</p><p><strong>Conclusion: </strong>We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"164"},"PeriodicalIF":0.0000,"publicationDate":"2020-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01094-y","citationCount":"7","resultStr":"{\"title\":\"Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly- a case report.\",\"authors\":\"D Hettiarachchi, Hetalkumar Panchal, P S Lai, V H W Dissanayake\",\"doi\":\"10.1186/s12881-020-01094-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.</p><p><strong>Case presentation: </strong>The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).</p><p><strong>Conclusion: </strong>We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"164\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-08-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01094-y\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01094-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01094-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 7
摘要
背景:先天性半发育不良伴鱼鳞状红皮病和肢体缺陷,也称为CHILD综合征,是一种x连锁显性、男性致死性遗传性皮肤病,患病率为10万分之一。位于Xq28的NSDHL基因突变可能损害NAD(P) H类固醇脱氢酶样蛋白的功能,并可能导致其发病。病例介绍:先证者是一名9个月大的双胞胎(T2)女孩,其健康的双胞胎姐妹(T1)是由非近亲父母所生的斯里兰卡裔。自出生以来,她表现为右侧持续的鱼鳞样红皮病和同侧肢体缺陷和先天性半发育不良。值得注意的是,患儿有同侧手发育不全和并指畸形。还有其他内脏异常。我们进行了全外显子组测序,发现了一个新的杂合变异(NSDHL, c.713C > a, p.Thr238Asn)。结论:我们报道了一种位于高度保守区域的NSDHL基因的新错义变异。这种变异通过减少活性位点的数量影响NAD(P) H类固醇脱氢酶样蛋白的功能,从而导致CHILD综合征表型和并指畸形。
Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly- a case report.
Background: Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.
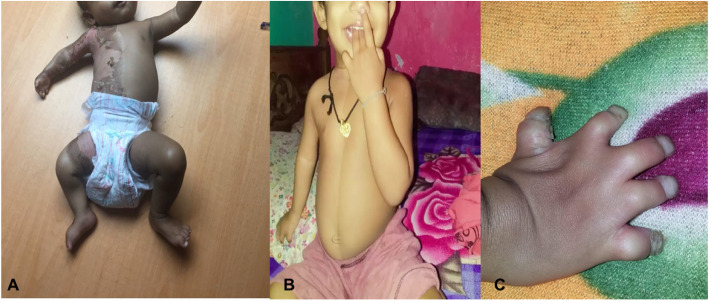
Case presentation: The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).
Conclusion: We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
