{"title":"全外显子组测序鉴定了与Snyder-Robinson综合征相关的精胺合成酶基因(SMS)的新突变。","authors":"Talal J Qazi, Qiao Wu, Ailikemu Aierken, Daru Lu, Ihtisham Bukhari, Hafiz M J Hussain, Jingmin Yang, Asif Mir, Hong Qing","doi":"10.1186/s12881-020-01095-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Loss of function mutations in the spermine synthase gene (SMS) have been reported to cause a rare X-linked intellectual disability known as Snyder-Robinson Syndrome (SRS). Besides intellectual disability, SRS is also characterized by reduced bone density, osteoporosis and facial dysmorphism. SRS phenotypes evolve with age from childhood to adulthood.</p><p><strong>Methods: </strong>Whole exome sequencing was performed to know the causative gene/pathogenic variant. Later we confirmed the pathogenic variant through Sanger sequencing. Furthermore, we also performed the mutational analysis through HOPE SERVER and SWISS-MODEL. Also, radiographs were also obtained for affected individual to confirm the disease features.</p><p><strong>Results: </strong>In this article, we report the first Pakistani family consisting of three patients with SRS and a novel missense pathogenic variant in the SMS gene (c.905 C > T p.(Ser302Leu)). In addition to the typical phenotypes, one patient presented with early-onset seizures. Clinical features, genetic and in-silico analysis linked the affected patients of the family with Snyder-Robinson and suggest that this novel mutation affects the spermine synthase activity.</p><p><strong>Conclusion: </strong>A novel missense variant in the SMS, c.905C > T p. (Ser302Leu), causing Snyder- Robinson Syndrome (SRS) is reported in three members of Pakistani Family.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"168"},"PeriodicalIF":0.0000,"publicationDate":"2020-08-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01095-x","citationCount":"1","resultStr":"{\"title\":\"Whole-exome sequencing identifies a novel mutation in spermine synthase gene (SMS) associated with Snyder-Robinson Syndrome.\",\"authors\":\"Talal J Qazi, Qiao Wu, Ailikemu Aierken, Daru Lu, Ihtisham Bukhari, Hafiz M J Hussain, Jingmin Yang, Asif Mir, Hong Qing\",\"doi\":\"10.1186/s12881-020-01095-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Loss of function mutations in the spermine synthase gene (SMS) have been reported to cause a rare X-linked intellectual disability known as Snyder-Robinson Syndrome (SRS). Besides intellectual disability, SRS is also characterized by reduced bone density, osteoporosis and facial dysmorphism. SRS phenotypes evolve with age from childhood to adulthood.</p><p><strong>Methods: </strong>Whole exome sequencing was performed to know the causative gene/pathogenic variant. Later we confirmed the pathogenic variant through Sanger sequencing. Furthermore, we also performed the mutational analysis through HOPE SERVER and SWISS-MODEL. Also, radiographs were also obtained for affected individual to confirm the disease features.</p><p><strong>Results: </strong>In this article, we report the first Pakistani family consisting of three patients with SRS and a novel missense pathogenic variant in the SMS gene (c.905 C > T p.(Ser302Leu)). In addition to the typical phenotypes, one patient presented with early-onset seizures. Clinical features, genetic and in-silico analysis linked the affected patients of the family with Snyder-Robinson and suggest that this novel mutation affects the spermine synthase activity.</p><p><strong>Conclusion: </strong>A novel missense variant in the SMS, c.905C > T p. (Ser302Leu), causing Snyder- Robinson Syndrome (SRS) is reported in three members of Pakistani Family.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"168\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-08-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01095-x\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01095-x\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01095-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 1
摘要
背景:精胺合酶基因(SMS)的功能突变丧失已被报道导致一种罕见的x连锁智力残疾,称为Snyder-Robinson综合征(SRS)。除智力残疾外,SRS还表现为骨密度降低、骨质疏松和面部畸形。从童年到成年,SRS表型随着年龄的增长而进化。方法:采用全外显子组测序法确定致病基因/致病变异。后来我们通过桑格测序确认了致病变异。此外,我们还通过HOPE SERVER和SWISS-MODEL进行了突变分析。此外,还对受影响的个体进行x线片检查以确认疾病特征。结果:在这篇文章中,我们报道了巴基斯坦第一个由三名SRS患者组成的家庭和一种新的SMS基因错义致病变异(c.905)C > p.(Ser302Leu))。除了典型的表型外,一名患者出现早发性癫痫发作。临床特征、遗传和计算机分析将该家族的受影响患者与Snyder-Robinson联系起来,并表明这种新的突变影响精胺合酶活性。结论:在巴基斯坦家族3名成员中发现了一种新的missense变异,c.905C > T . p. (Ser302Leu),可引起Snyder- Robinson综合征(SRS)。
Whole-exome sequencing identifies a novel mutation in spermine synthase gene (SMS) associated with Snyder-Robinson Syndrome.
Background: Loss of function mutations in the spermine synthase gene (SMS) have been reported to cause a rare X-linked intellectual disability known as Snyder-Robinson Syndrome (SRS). Besides intellectual disability, SRS is also characterized by reduced bone density, osteoporosis and facial dysmorphism. SRS phenotypes evolve with age from childhood to adulthood.
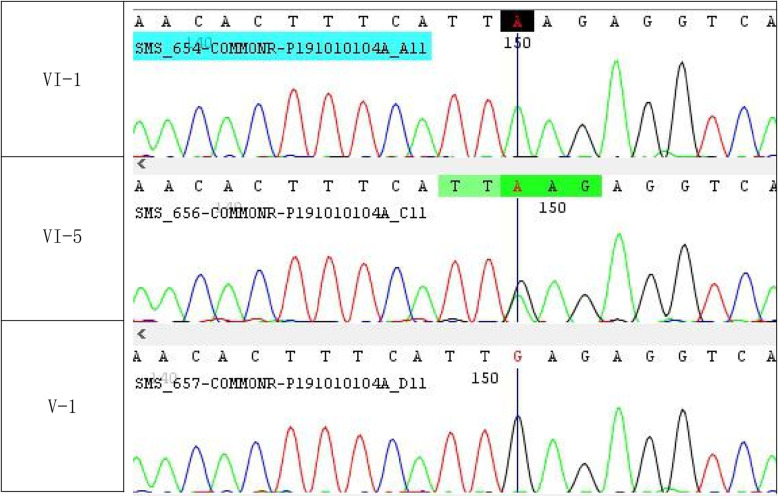
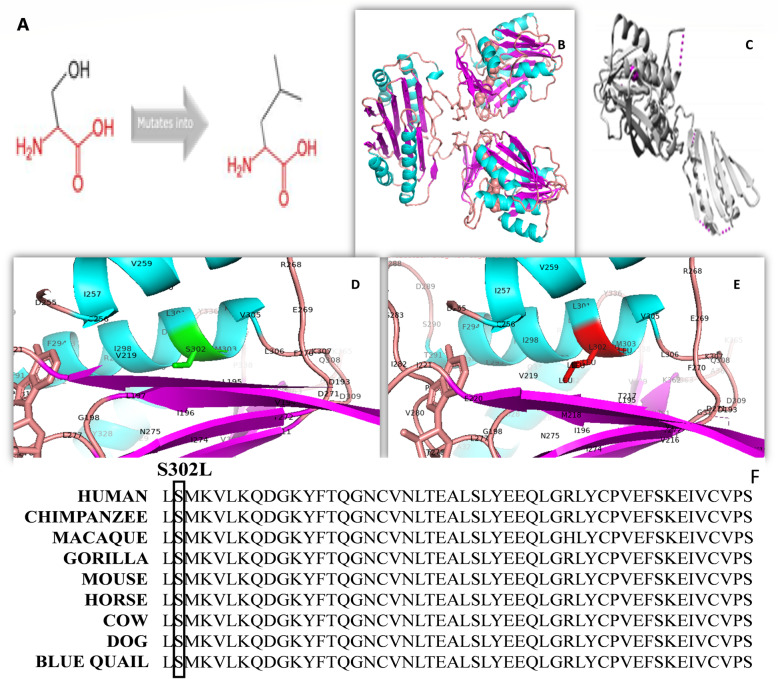
Methods: Whole exome sequencing was performed to know the causative gene/pathogenic variant. Later we confirmed the pathogenic variant through Sanger sequencing. Furthermore, we also performed the mutational analysis through HOPE SERVER and SWISS-MODEL. Also, radiographs were also obtained for affected individual to confirm the disease features.
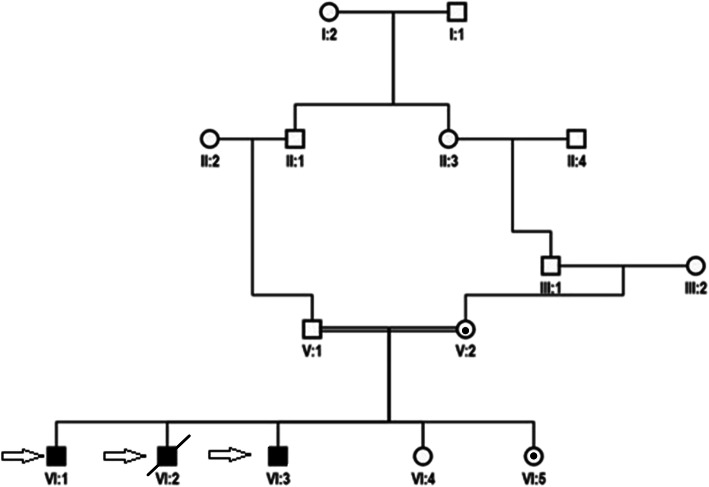
Results: In this article, we report the first Pakistani family consisting of three patients with SRS and a novel missense pathogenic variant in the SMS gene (c.905 C > T p.(Ser302Leu)). In addition to the typical phenotypes, one patient presented with early-onset seizures. Clinical features, genetic and in-silico analysis linked the affected patients of the family with Snyder-Robinson and suggest that this novel mutation affects the spermine synthase activity.
Conclusion: A novel missense variant in the SMS, c.905C > T p. (Ser302Leu), causing Snyder- Robinson Syndrome (SRS) is reported in three members of Pakistani Family.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
