{"title":"一例痉挛性麻痹患者在发病9年后最终被诊断为V180I遗传性克雅氏病。","authors":"Taichi Nomura, Ikuko Iwata, Ryoji Naganuma, Masaaki Matsushima, Katsuya Satoh, Tetsuyuki Kitamoto, Ichiro Yabe","doi":"10.1080/19336896.2020.1823179","DOIUrl":null,"url":null,"abstract":"ABSTRACT Genetic Creutzfeldt-Jakob disease (gCJD) with a mutation in codon 180 of the prion protein gene (V180I gCJD) is the most common form of gCJD in Japan, but only a few cases have been reported in Europe and the United States. It is clinically characterized by occurring in the elderly and presenting as slowly progressive dementia, although it generally shows less cerebellar and pyramidal symptoms than sporadic CJD. Here, we report a patient with V180I gCJD who initially presented with slowly progressive spastic paralysis with neither cerebrospinal fluid (CSF) nor magnetic resonance imaging (MRI) abnormalities. His symptoms progressed gradually, and after 9 years, he displayed features more typical of CJD. Diffusion-weighted MRI revealed high-intensity signals in the cortical gyrus, and there was a marked increase of 14-3-3 protein and total tau protein in the CSF, but he was negative for the real-time quaking-induced conversion assay. Although the time course was more consistent with Gerstmann-Sträussler-Scheinker disease than CJD, genetic testing revealed V180I gCJD. This is the first report of a patient with V180I gCJD who initially presented with spastic paralysis, and also the first to reveal that it took 9 years from disease onset for cortical dysfunction to develop and for MRI and CSF abnormalities to be detectable. In conclusion, we should screen for V180I gCJD in elderly patients presenting with slowly progressive spastic paralysis.","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":" ","pages":"226-231"},"PeriodicalIF":17.7000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/19336896.2020.1823179","citationCount":"3","resultStr":"{\"title\":\"A patient with spastic paralysis finally diagnosed as V180I genetic Creutzfeldt-Jakob disease 9 years after onset.\",\"authors\":\"Taichi Nomura, Ikuko Iwata, Ryoji Naganuma, Masaaki Matsushima, Katsuya Satoh, Tetsuyuki Kitamoto, Ichiro Yabe\",\"doi\":\"10.1080/19336896.2020.1823179\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"ABSTRACT Genetic Creutzfeldt-Jakob disease (gCJD) with a mutation in codon 180 of the prion protein gene (V180I gCJD) is the most common form of gCJD in Japan, but only a few cases have been reported in Europe and the United States. It is clinically characterized by occurring in the elderly and presenting as slowly progressive dementia, although it generally shows less cerebellar and pyramidal symptoms than sporadic CJD. Here, we report a patient with V180I gCJD who initially presented with slowly progressive spastic paralysis with neither cerebrospinal fluid (CSF) nor magnetic resonance imaging (MRI) abnormalities. His symptoms progressed gradually, and after 9 years, he displayed features more typical of CJD. Diffusion-weighted MRI revealed high-intensity signals in the cortical gyrus, and there was a marked increase of 14-3-3 protein and total tau protein in the CSF, but he was negative for the real-time quaking-induced conversion assay. Although the time course was more consistent with Gerstmann-Sträussler-Scheinker disease than CJD, genetic testing revealed V180I gCJD. This is the first report of a patient with V180I gCJD who initially presented with spastic paralysis, and also the first to reveal that it took 9 years from disease onset for cortical dysfunction to develop and for MRI and CSF abnormalities to be detectable. In conclusion, we should screen for V180I gCJD in elderly patients presenting with slowly progressive spastic paralysis.\",\"PeriodicalId\":1,\"journal\":{\"name\":\"Accounts of Chemical Research\",\"volume\":\" \",\"pages\":\"226-231\"},\"PeriodicalIF\":17.7000,\"publicationDate\":\"2020-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1080/19336896.2020.1823179\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Accounts of Chemical Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1080/19336896.2020.1823179\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1080/19336896.2020.1823179","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
A patient with spastic paralysis finally diagnosed as V180I genetic Creutzfeldt-Jakob disease 9 years after onset.
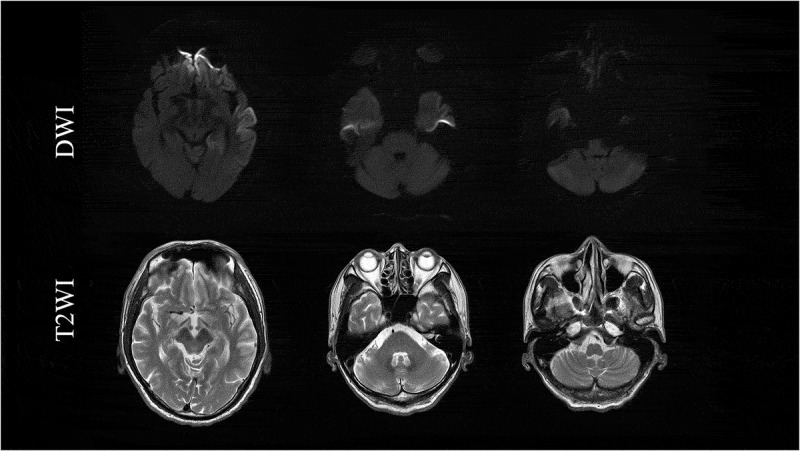
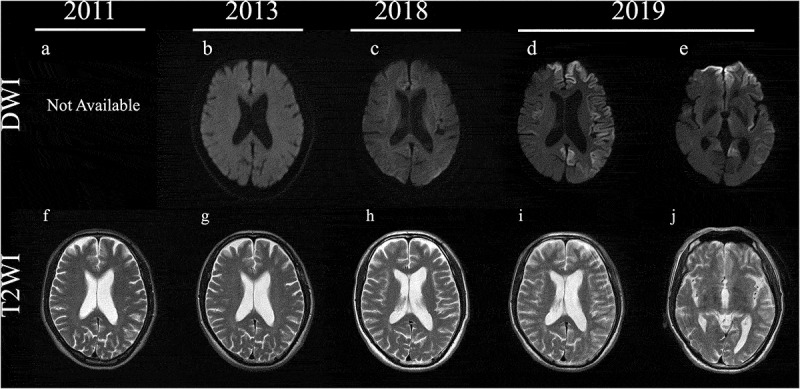
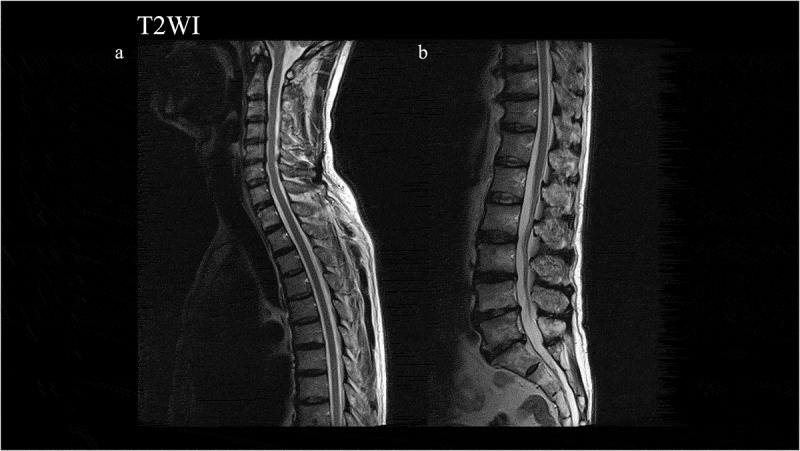
ABSTRACT Genetic Creutzfeldt-Jakob disease (gCJD) with a mutation in codon 180 of the prion protein gene (V180I gCJD) is the most common form of gCJD in Japan, but only a few cases have been reported in Europe and the United States. It is clinically characterized by occurring in the elderly and presenting as slowly progressive dementia, although it generally shows less cerebellar and pyramidal symptoms than sporadic CJD. Here, we report a patient with V180I gCJD who initially presented with slowly progressive spastic paralysis with neither cerebrospinal fluid (CSF) nor magnetic resonance imaging (MRI) abnormalities. His symptoms progressed gradually, and after 9 years, he displayed features more typical of CJD. Diffusion-weighted MRI revealed high-intensity signals in the cortical gyrus, and there was a marked increase of 14-3-3 protein and total tau protein in the CSF, but he was negative for the real-time quaking-induced conversion assay. Although the time course was more consistent with Gerstmann-Sträussler-Scheinker disease than CJD, genetic testing revealed V180I gCJD. This is the first report of a patient with V180I gCJD who initially presented with spastic paralysis, and also the first to reveal that it took 9 years from disease onset for cortical dysfunction to develop and for MRI and CSF abnormalities to be detectable. In conclusion, we should screen for V180I gCJD in elderly patients presenting with slowly progressive spastic paralysis.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
