Š Stangler Herodež, N Marčun Varda, Kokalj Vokač N, D Krgović
{"title":"新生儿先天性心脏异常的新生KMT2D杂合移码缺失。","authors":"Š Stangler Herodež, N Marčun Varda, Kokalj Vokač N, D Krgović","doi":"10.2478/bjmg-2020-0008","DOIUrl":null,"url":null,"abstract":"<p><p>Kabuki syndrome (KS) is characterized by typical facial features and patients are also affected by multiple congenital anomalies, of which congenital heart anomalies (CHAs) are present in 28.0 to 80.0%. In approximately 75.0% of patients, the genetic causes of KS are caused by mutation in the <i>KMT2D</i> gene. Although KS is a well-characterized syndrome, reaching the diagnosis in neonates is still challenging. Namely, newborns usually display mild facial features; therefore the diagnosis is mainly based on congenital malformations. In our case, a newborn was referred for next generation sequencing (NGS) testing due to the prenatally observed CHA. After birth, a ventricular septal defect (VSD), vesicoureteral reflux, muscular hypotonia, cleft palate, mild microcephaly, and some dysmorphic features, were noted. The NGS analysis was performed on the proband's genomic DNA using the TruSight One Sequencing Panel, which enriches exons of 4813 genes with clinical relevance to the disease. After variant calling, NGS data analysis was predominantly focused on rare variants in genes involved in VSD, microcephaly, and muscular hypotonia; features observed predominantly in our proband. With the aforementioned protocol, we were able to determine the previously unreported <i>de novo</i> frameshift deletion in the <i>KMT2D</i> gene resulting in translation termination. Although our proband is a typical representative of KS, his diagnosis was reached only after NGS analysis. Our proband thus represents the importance of genotypephenotype driven NGS analysis in diagnosis of patients with congenital anomalies.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"23 1","pages":"83-90"},"PeriodicalIF":0.9000,"publicationDate":"2020-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d7/85/bjmg-23-083.PMC7474217.pdf","citationCount":"7","resultStr":"{\"title\":\"<i>De Novo KMT2D</i> Heterozygous Frameshift Deletion in a Newborn with a Congenital Heart Anomaly.\",\"authors\":\"Š Stangler Herodež, N Marčun Varda, Kokalj Vokač N, D Krgović\",\"doi\":\"10.2478/bjmg-2020-0008\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Kabuki syndrome (KS) is characterized by typical facial features and patients are also affected by multiple congenital anomalies, of which congenital heart anomalies (CHAs) are present in 28.0 to 80.0%. In approximately 75.0% of patients, the genetic causes of KS are caused by mutation in the <i>KMT2D</i> gene. Although KS is a well-characterized syndrome, reaching the diagnosis in neonates is still challenging. Namely, newborns usually display mild facial features; therefore the diagnosis is mainly based on congenital malformations. In our case, a newborn was referred for next generation sequencing (NGS) testing due to the prenatally observed CHA. After birth, a ventricular septal defect (VSD), vesicoureteral reflux, muscular hypotonia, cleft palate, mild microcephaly, and some dysmorphic features, were noted. The NGS analysis was performed on the proband's genomic DNA using the TruSight One Sequencing Panel, which enriches exons of 4813 genes with clinical relevance to the disease. After variant calling, NGS data analysis was predominantly focused on rare variants in genes involved in VSD, microcephaly, and muscular hypotonia; features observed predominantly in our proband. With the aforementioned protocol, we were able to determine the previously unreported <i>de novo</i> frameshift deletion in the <i>KMT2D</i> gene resulting in translation termination. Although our proband is a typical representative of KS, his diagnosis was reached only after NGS analysis. Our proband thus represents the importance of genotypephenotype driven NGS analysis in diagnosis of patients with congenital anomalies.</p>\",\"PeriodicalId\":55403,\"journal\":{\"name\":\"Balkan Journal of Medical Genetics\",\"volume\":\"23 1\",\"pages\":\"83-90\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2020-08-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d7/85/bjmg-23-083.PMC7474217.pdf\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Balkan Journal of Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2478/bjmg-2020-0008\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/6/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2020-0008","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/6/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
De Novo KMT2D Heterozygous Frameshift Deletion in a Newborn with a Congenital Heart Anomaly.
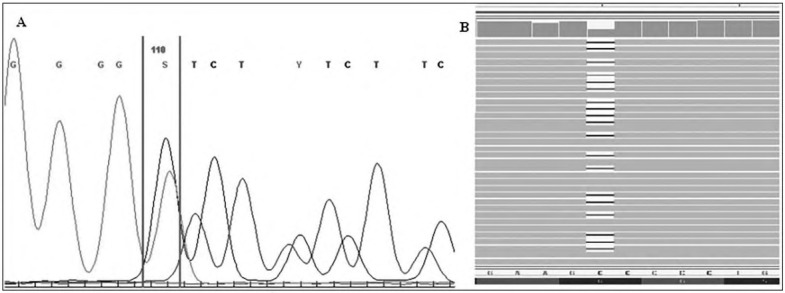
Kabuki syndrome (KS) is characterized by typical facial features and patients are also affected by multiple congenital anomalies, of which congenital heart anomalies (CHAs) are present in 28.0 to 80.0%. In approximately 75.0% of patients, the genetic causes of KS are caused by mutation in the KMT2D gene. Although KS is a well-characterized syndrome, reaching the diagnosis in neonates is still challenging. Namely, newborns usually display mild facial features; therefore the diagnosis is mainly based on congenital malformations. In our case, a newborn was referred for next generation sequencing (NGS) testing due to the prenatally observed CHA. After birth, a ventricular septal defect (VSD), vesicoureteral reflux, muscular hypotonia, cleft palate, mild microcephaly, and some dysmorphic features, were noted. The NGS analysis was performed on the proband's genomic DNA using the TruSight One Sequencing Panel, which enriches exons of 4813 genes with clinical relevance to the disease. After variant calling, NGS data analysis was predominantly focused on rare variants in genes involved in VSD, microcephaly, and muscular hypotonia; features observed predominantly in our proband. With the aforementioned protocol, we were able to determine the previously unreported de novo frameshift deletion in the KMT2D gene resulting in translation termination. Although our proband is a typical representative of KS, his diagnosis was reached only after NGS analysis. Our proband thus represents the importance of genotypephenotype driven NGS analysis in diagnosis of patients with congenital anomalies.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
