Katina Kartalias, Austin P Gillies, Maria T Peña, Andrea Estrada, Dorothy I Bulas, Carlos R Ferreira, Laura L Tosi
{"title":"对一名患肢端骨发育不良儿童的14年随访,强调多学科治疗的必要性:一份病例报告。","authors":"Katina Kartalias, Austin P Gillies, Maria T Peña, Andrea Estrada, Dorothy I Bulas, Carlos R Ferreira, Laura L Tosi","doi":"10.1186/s12881-020-01127-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Acroscyphodysplasia has been described as a phenotypic variant of acrodysostosis type 2 and pseudohypoparathyroidism. In acrodysostosis, skeletal features can include brachydactyly, facial hypoplasia, cone-shaped epiphyses, short stature, and advanced bone age. To date, reports on this disorder have focused on phenotypic findings, endocrine changes, and genetic variation. We present a 14-year overview of a patient, from birth to skeletal maturity, with acroscyphodysplasia, noting the significant orthopaedic challenges and the need for a multidisciplinary team, including specialists in genetics, orthopaedics, endocrinology, and otolaryngology, to optimize long-term outcomes.</p><p><strong>Case presentation: </strong>The patient presented as a newborn with dysmorphic facial features, including severe midface hypoplasia, malar flattening, nasal stenosis, and feeding difficulties. Radiologic findings were initially subtle, and a skeletal survey performed at age 7 months was initially considered normal. Genetic evaluation revealed a variant in PDE4D and subsequent pseudohypoparathyroidism. The patient presented to the department of orthopaedics, at age 2 years 9 months with a leg length discrepancy, right knee contracture, and severely crouched gait. Radiographs demonstrated cone-shaped epiphyses of the right distal femur and proximal tibia, but no evidence of growth plate changes in the left leg. The child developed early posterior epiphyseal arrest on the right side and required multiple surgical interventions to achieve neutral extension. Her left distal femur developed late posterior physeal arrest and secondary contracture without evidence of schypho deformity, which improved with anterior screw epiphysiodesis. The child required numerous orthopaedic surgical interventions to achieve full knee extension bilaterally. At age 13 years 11 months, she was an independent ambulator with erect posture. The child underwent numerous otolaryngology procedures and will require significant ongoing care. She has moderate intellectual disability.</p><p><strong>Discussion and conclusions: </strong>Key challenges in the management of this case included the subtle changes on initial skeletal survey and the marked asymmetry of her deformity. While cone-shaped epiphyses are a hallmark of acrodysostosis, posterior tethering/growth arrest of the posterior distal femur has not been previously reported. Correction of the secondary knee contracture was essential to improve ambulation. Children with acroscyphodysplasia require a multidisciplinary approach, including radiology, genetics, orthopaedics, otolaryngology, and endocrinology specialties.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"189"},"PeriodicalIF":0.0000,"publicationDate":"2020-09-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12881-020-01127-6","citationCount":"2","resultStr":"{\"title\":\"Fourteen-year follow-up of a child with acroscyphodysplasia with emphasis on the need for multidisciplinary management: a case report.\",\"authors\":\"Katina Kartalias, Austin P Gillies, Maria T Peña, Andrea Estrada, Dorothy I Bulas, Carlos R Ferreira, Laura L Tosi\",\"doi\":\"10.1186/s12881-020-01127-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Acroscyphodysplasia has been described as a phenotypic variant of acrodysostosis type 2 and pseudohypoparathyroidism. In acrodysostosis, skeletal features can include brachydactyly, facial hypoplasia, cone-shaped epiphyses, short stature, and advanced bone age. To date, reports on this disorder have focused on phenotypic findings, endocrine changes, and genetic variation. We present a 14-year overview of a patient, from birth to skeletal maturity, with acroscyphodysplasia, noting the significant orthopaedic challenges and the need for a multidisciplinary team, including specialists in genetics, orthopaedics, endocrinology, and otolaryngology, to optimize long-term outcomes.</p><p><strong>Case presentation: </strong>The patient presented as a newborn with dysmorphic facial features, including severe midface hypoplasia, malar flattening, nasal stenosis, and feeding difficulties. Radiologic findings were initially subtle, and a skeletal survey performed at age 7 months was initially considered normal. Genetic evaluation revealed a variant in PDE4D and subsequent pseudohypoparathyroidism. The patient presented to the department of orthopaedics, at age 2 years 9 months with a leg length discrepancy, right knee contracture, and severely crouched gait. Radiographs demonstrated cone-shaped epiphyses of the right distal femur and proximal tibia, but no evidence of growth plate changes in the left leg. The child developed early posterior epiphyseal arrest on the right side and required multiple surgical interventions to achieve neutral extension. Her left distal femur developed late posterior physeal arrest and secondary contracture without evidence of schypho deformity, which improved with anterior screw epiphysiodesis. The child required numerous orthopaedic surgical interventions to achieve full knee extension bilaterally. At age 13 years 11 months, she was an independent ambulator with erect posture. The child underwent numerous otolaryngology procedures and will require significant ongoing care. She has moderate intellectual disability.</p><p><strong>Discussion and conclusions: </strong>Key challenges in the management of this case included the subtle changes on initial skeletal survey and the marked asymmetry of her deformity. While cone-shaped epiphyses are a hallmark of acrodysostosis, posterior tethering/growth arrest of the posterior distal femur has not been previously reported. Correction of the secondary knee contracture was essential to improve ambulation. Children with acroscyphodysplasia require a multidisciplinary approach, including radiology, genetics, orthopaedics, otolaryngology, and endocrinology specialties.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"189\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-09-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12881-020-01127-6\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01127-6\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01127-6","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
Fourteen-year follow-up of a child with acroscyphodysplasia with emphasis on the need for multidisciplinary management: a case report.
Background: Acroscyphodysplasia has been described as a phenotypic variant of acrodysostosis type 2 and pseudohypoparathyroidism. In acrodysostosis, skeletal features can include brachydactyly, facial hypoplasia, cone-shaped epiphyses, short stature, and advanced bone age. To date, reports on this disorder have focused on phenotypic findings, endocrine changes, and genetic variation. We present a 14-year overview of a patient, from birth to skeletal maturity, with acroscyphodysplasia, noting the significant orthopaedic challenges and the need for a multidisciplinary team, including specialists in genetics, orthopaedics, endocrinology, and otolaryngology, to optimize long-term outcomes.
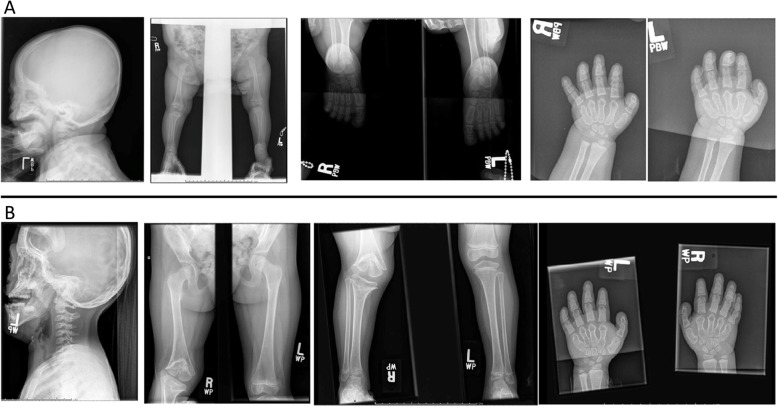
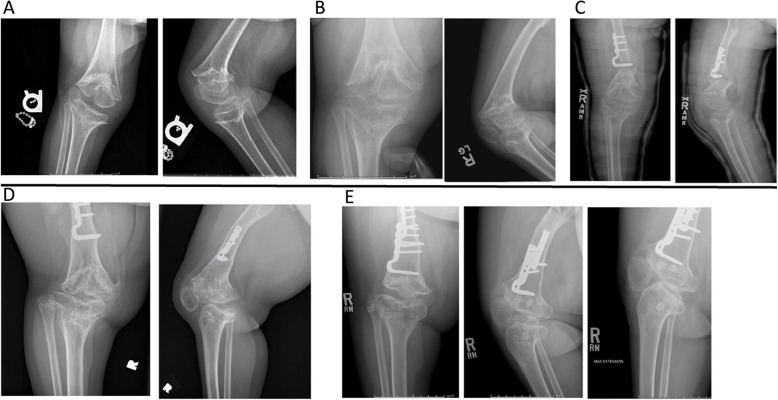
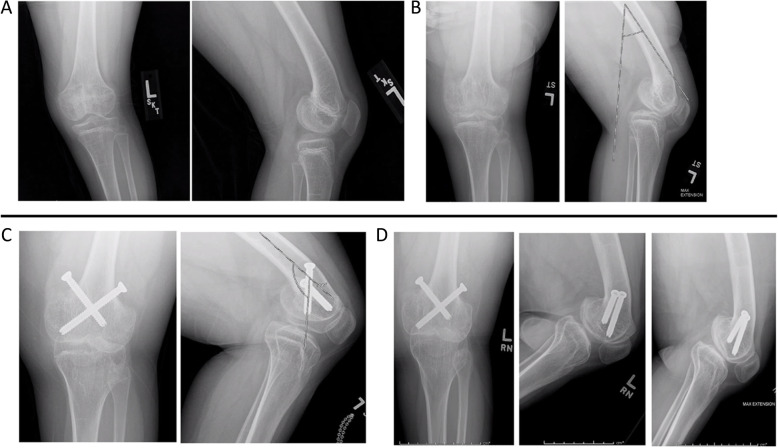
Case presentation: The patient presented as a newborn with dysmorphic facial features, including severe midface hypoplasia, malar flattening, nasal stenosis, and feeding difficulties. Radiologic findings were initially subtle, and a skeletal survey performed at age 7 months was initially considered normal. Genetic evaluation revealed a variant in PDE4D and subsequent pseudohypoparathyroidism. The patient presented to the department of orthopaedics, at age 2 years 9 months with a leg length discrepancy, right knee contracture, and severely crouched gait. Radiographs demonstrated cone-shaped epiphyses of the right distal femur and proximal tibia, but no evidence of growth plate changes in the left leg. The child developed early posterior epiphyseal arrest on the right side and required multiple surgical interventions to achieve neutral extension. Her left distal femur developed late posterior physeal arrest and secondary contracture without evidence of schypho deformity, which improved with anterior screw epiphysiodesis. The child required numerous orthopaedic surgical interventions to achieve full knee extension bilaterally. At age 13 years 11 months, she was an independent ambulator with erect posture. The child underwent numerous otolaryngology procedures and will require significant ongoing care. She has moderate intellectual disability.
Discussion and conclusions: Key challenges in the management of this case included the subtle changes on initial skeletal survey and the marked asymmetry of her deformity. While cone-shaped epiphyses are a hallmark of acrodysostosis, posterior tethering/growth arrest of the posterior distal femur has not been previously reported. Correction of the secondary knee contracture was essential to improve ambulation. Children with acroscyphodysplasia require a multidisciplinary approach, including radiology, genetics, orthopaedics, otolaryngology, and endocrinology specialties.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
