{"title":"Steganinae果蝇(双翅目:果蝇科)的系统基因组研究:强大的基因树异质性和单系性证据。","authors":"Guilherme Rezende Dias, Eduardo Guimarães Dupim, Thyago Vanderlinde, Beatriz Mello, Antonio Bernardo Carvalho","doi":"10.1186/s12862-020-01703-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The Drosophilidae family is traditionally divided into two subfamilies: Drosophilinae and Steganinae. This division is based on morphological characters, and the two subfamilies have been treated as monophyletic in most of the literature, but some molecular phylogenies have suggested Steganinae to be paraphyletic. To test the paraphyletic-Steganinae hypothesis, here, we used genomic sequences of eight Drosophilidae (three Steganinae and five Drosophilinae) and two Ephydridae (outgroup) species and inferred the phylogeny for the group based on a dataset of 1,028 orthologous genes present in all species (> 1,000,000 bp). This dataset includes three genera that broke the monophyly of the subfamilies in previous works. To investigate possible biases introduced by small sample sizes and automatic gene annotation, we used the same methods to infer species trees from a set of 10 manually annotated genes that are commonly used in phylogenetics.</p><p><strong>Results: </strong>Most of the 1,028 gene trees depicted Steganinae as paraphyletic with distinct topologies, but the most common topology depicted it as monophyletic (43.7% of the gene trees). Despite the high levels of gene tree heterogeneity observed, species tree inference in ASTRAL, in PhyloNet, and with the concatenation approach strongly supported the monophyly of both subfamilies for the 1,028-gene dataset. However, when using the concatenation approach to infer a species tree from the smaller set of 10 genes, we recovered Steganinae as a paraphyletic group. The pattern of gene tree heterogeneity was asymmetrical and thus could not be explained solely by incomplete lineage sorting (ILS).</p><p><strong>Conclusions: </strong>Steganinae was clearly a monophyletic group in the dataset that we analyzed. In addition to ILS, gene tree discordance was possibly the result of introgression, suggesting complex branching processes during the early evolution of Drosophilidae with short speciation intervals and gene flow. Our study highlights the importance of genomic data in elucidating contentious phylogenetic relationships and suggests that phylogenetic inference for drosophilids based on small molecular datasets should be performed cautiously. Finally, we suggest an approach for the correction and cleaning of BUSCO-derived genomic datasets that will be useful to other researchers planning to use this tool for phylogenomic studies.</p>","PeriodicalId":9111,"journal":{"name":"BMC Evolutionary Biology","volume":"20 1","pages":"141"},"PeriodicalIF":3.4000,"publicationDate":"2020-11-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7607883/pdf/","citationCount":"0","resultStr":"{\"title\":\"A phylogenomic study of Steganinae fruit flies (Diptera: Drosophilidae): strong gene tree heterogeneity and evidence for monophyly.\",\"authors\":\"Guilherme Rezende Dias, Eduardo Guimarães Dupim, Thyago Vanderlinde, Beatriz Mello, Antonio Bernardo Carvalho\",\"doi\":\"10.1186/s12862-020-01703-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The Drosophilidae family is traditionally divided into two subfamilies: Drosophilinae and Steganinae. This division is based on morphological characters, and the two subfamilies have been treated as monophyletic in most of the literature, but some molecular phylogenies have suggested Steganinae to be paraphyletic. To test the paraphyletic-Steganinae hypothesis, here, we used genomic sequences of eight Drosophilidae (three Steganinae and five Drosophilinae) and two Ephydridae (outgroup) species and inferred the phylogeny for the group based on a dataset of 1,028 orthologous genes present in all species (> 1,000,000 bp). This dataset includes three genera that broke the monophyly of the subfamilies in previous works. To investigate possible biases introduced by small sample sizes and automatic gene annotation, we used the same methods to infer species trees from a set of 10 manually annotated genes that are commonly used in phylogenetics.</p><p><strong>Results: </strong>Most of the 1,028 gene trees depicted Steganinae as paraphyletic with distinct topologies, but the most common topology depicted it as monophyletic (43.7% of the gene trees). Despite the high levels of gene tree heterogeneity observed, species tree inference in ASTRAL, in PhyloNet, and with the concatenation approach strongly supported the monophyly of both subfamilies for the 1,028-gene dataset. However, when using the concatenation approach to infer a species tree from the smaller set of 10 genes, we recovered Steganinae as a paraphyletic group. The pattern of gene tree heterogeneity was asymmetrical and thus could not be explained solely by incomplete lineage sorting (ILS).</p><p><strong>Conclusions: </strong>Steganinae was clearly a monophyletic group in the dataset that we analyzed. In addition to ILS, gene tree discordance was possibly the result of introgression, suggesting complex branching processes during the early evolution of Drosophilidae with short speciation intervals and gene flow. Our study highlights the importance of genomic data in elucidating contentious phylogenetic relationships and suggests that phylogenetic inference for drosophilids based on small molecular datasets should be performed cautiously. Finally, we suggest an approach for the correction and cleaning of BUSCO-derived genomic datasets that will be useful to other researchers planning to use this tool for phylogenomic studies.</p>\",\"PeriodicalId\":9111,\"journal\":{\"name\":\"BMC Evolutionary Biology\",\"volume\":\"20 1\",\"pages\":\"141\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2020-11-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7607883/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Evolutionary Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12862-020-01703-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Evolutionary Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12862-020-01703-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
A phylogenomic study of Steganinae fruit flies (Diptera: Drosophilidae): strong gene tree heterogeneity and evidence for monophyly.
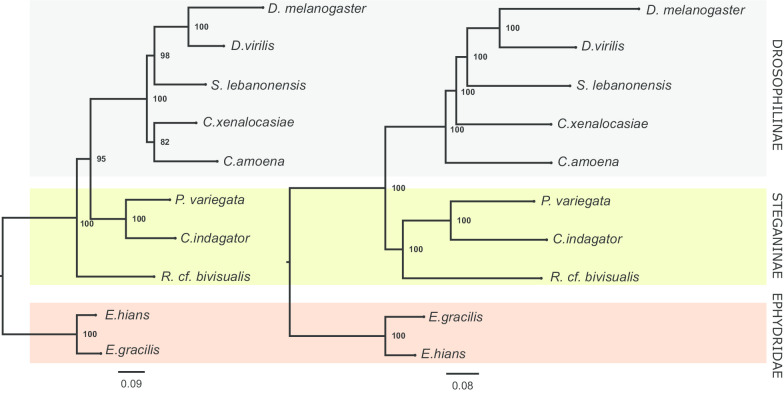
Background: The Drosophilidae family is traditionally divided into two subfamilies: Drosophilinae and Steganinae. This division is based on morphological characters, and the two subfamilies have been treated as monophyletic in most of the literature, but some molecular phylogenies have suggested Steganinae to be paraphyletic. To test the paraphyletic-Steganinae hypothesis, here, we used genomic sequences of eight Drosophilidae (three Steganinae and five Drosophilinae) and two Ephydridae (outgroup) species and inferred the phylogeny for the group based on a dataset of 1,028 orthologous genes present in all species (> 1,000,000 bp). This dataset includes three genera that broke the monophyly of the subfamilies in previous works. To investigate possible biases introduced by small sample sizes and automatic gene annotation, we used the same methods to infer species trees from a set of 10 manually annotated genes that are commonly used in phylogenetics.
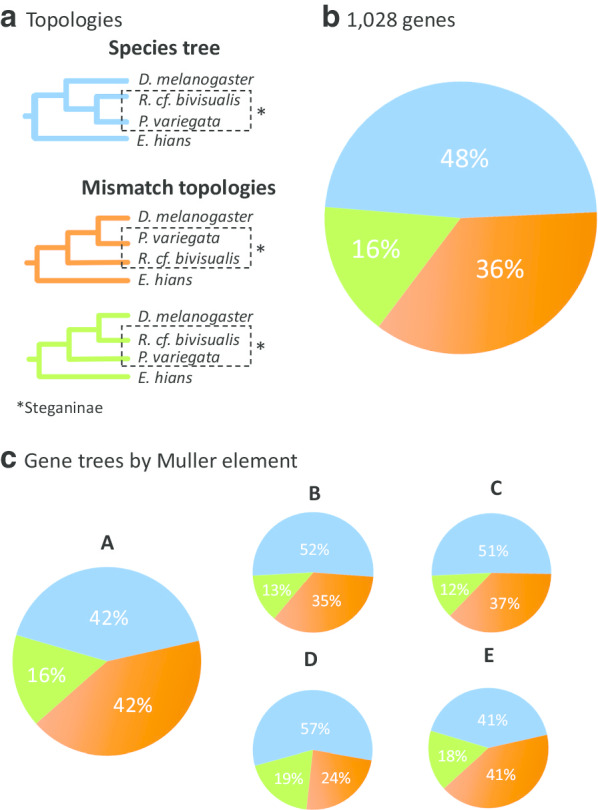
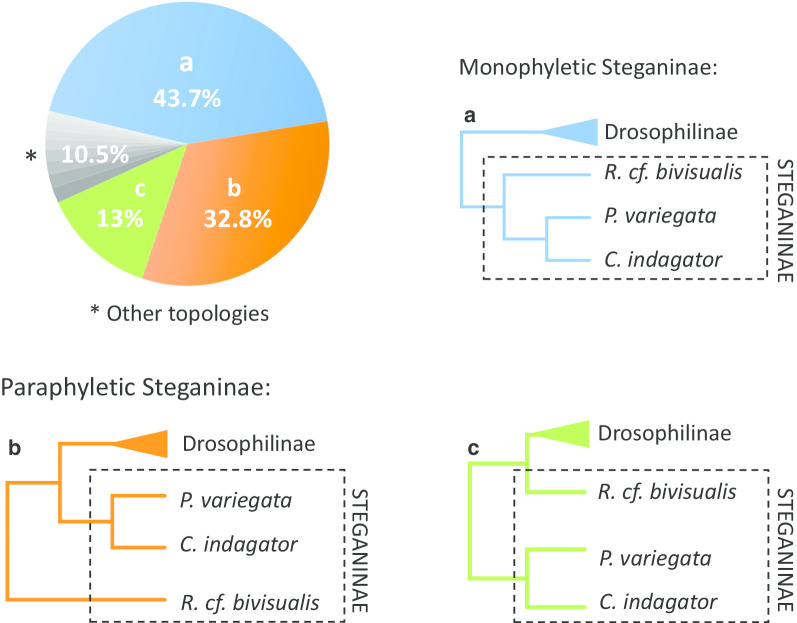
Results: Most of the 1,028 gene trees depicted Steganinae as paraphyletic with distinct topologies, but the most common topology depicted it as monophyletic (43.7% of the gene trees). Despite the high levels of gene tree heterogeneity observed, species tree inference in ASTRAL, in PhyloNet, and with the concatenation approach strongly supported the monophyly of both subfamilies for the 1,028-gene dataset. However, when using the concatenation approach to infer a species tree from the smaller set of 10 genes, we recovered Steganinae as a paraphyletic group. The pattern of gene tree heterogeneity was asymmetrical and thus could not be explained solely by incomplete lineage sorting (ILS).
Conclusions: Steganinae was clearly a monophyletic group in the dataset that we analyzed. In addition to ILS, gene tree discordance was possibly the result of introgression, suggesting complex branching processes during the early evolution of Drosophilidae with short speciation intervals and gene flow. Our study highlights the importance of genomic data in elucidating contentious phylogenetic relationships and suggests that phylogenetic inference for drosophilids based on small molecular datasets should be performed cautiously. Finally, we suggest an approach for the correction and cleaning of BUSCO-derived genomic datasets that will be useful to other researchers planning to use this tool for phylogenomic studies.
期刊介绍:
BMC Evolutionary Biology is an open access, peer-reviewed journal that considers articles on all aspects of molecular and non-molecular evolution of all organisms, as well as phylogenetics and palaeontology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
