{"title":"亨廷顿氏病和其他三重重复疾病的新进展:DNA修复转向阴暗面。","authors":"Robert S Lahue","doi":"10.1042/NS20200010","DOIUrl":null,"url":null,"abstract":"<p><p>Huntington's disease (HD) is a fatal, inherited neurodegenerative disease that causes neuronal death, particularly in medium spiny neurons. HD leads to serious and progressive motor, cognitive and psychiatric symptoms. Its genetic basis is an expansion of the CAG triplet repeat in the <i>HTT</i> gene, leading to extra glutamines in the huntingtin protein. HD is one of nine genetic diseases in this polyglutamine (polyQ) category, that also includes a number of inherited spinocerebellar ataxias (SCAs). Traditionally it has been assumed that HD age of onset and disease progression were solely the outcome of age-dependent exposure of neurons to toxic effects of the inherited mutant huntingtin protein. However, recent genome-wide association studies (GWAS) have revealed significant effects of genetic variants outside of <i>HTT</i>. Surprisingly, these variants turn out to be mostly in genes encoding DNA repair factors, suggesting that at least some disease modulation occurs at the level of the <i>HTT</i> DNA itself. These DNA repair proteins are known from model systems to promote ongoing somatic CAG repeat expansions in tissues affected by HD. Thus, for triplet repeats, some DNA repair proteins seem to abandon their normal genoprotective roles and, instead, drive expansions and accelerate disease. One attractive hypothesis-still to be proven rigorously-is that somatic <i>HTT</i> expansions augment the disease burden of the inherited allele. If so, therapeutic approaches that lower levels of huntingtin protein may need blending with additional therapies that reduce levels of somatic CAG repeat expansions to achieve maximal effect.</p>","PeriodicalId":74287,"journal":{"name":"Neuronal signaling","volume":"4 4","pages":"NS20200010"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7672267/pdf/","citationCount":"10","resultStr":"{\"title\":\"New developments in Huntington's disease and other triplet repeat diseases: DNA repair turns to the dark side.\",\"authors\":\"Robert S Lahue\",\"doi\":\"10.1042/NS20200010\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Huntington's disease (HD) is a fatal, inherited neurodegenerative disease that causes neuronal death, particularly in medium spiny neurons. HD leads to serious and progressive motor, cognitive and psychiatric symptoms. Its genetic basis is an expansion of the CAG triplet repeat in the <i>HTT</i> gene, leading to extra glutamines in the huntingtin protein. HD is one of nine genetic diseases in this polyglutamine (polyQ) category, that also includes a number of inherited spinocerebellar ataxias (SCAs). Traditionally it has been assumed that HD age of onset and disease progression were solely the outcome of age-dependent exposure of neurons to toxic effects of the inherited mutant huntingtin protein. However, recent genome-wide association studies (GWAS) have revealed significant effects of genetic variants outside of <i>HTT</i>. Surprisingly, these variants turn out to be mostly in genes encoding DNA repair factors, suggesting that at least some disease modulation occurs at the level of the <i>HTT</i> DNA itself. These DNA repair proteins are known from model systems to promote ongoing somatic CAG repeat expansions in tissues affected by HD. Thus, for triplet repeats, some DNA repair proteins seem to abandon their normal genoprotective roles and, instead, drive expansions and accelerate disease. One attractive hypothesis-still to be proven rigorously-is that somatic <i>HTT</i> expansions augment the disease burden of the inherited allele. If so, therapeutic approaches that lower levels of huntingtin protein may need blending with additional therapies that reduce levels of somatic CAG repeat expansions to achieve maximal effect.</p>\",\"PeriodicalId\":74287,\"journal\":{\"name\":\"Neuronal signaling\",\"volume\":\"4 4\",\"pages\":\"NS20200010\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-11-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7672267/pdf/\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neuronal signaling\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1042/NS20200010\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"Neuroscience\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuronal signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1042/NS20200010","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/12/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"Neuroscience","Score":null,"Total":0}
New developments in Huntington's disease and other triplet repeat diseases: DNA repair turns to the dark side.
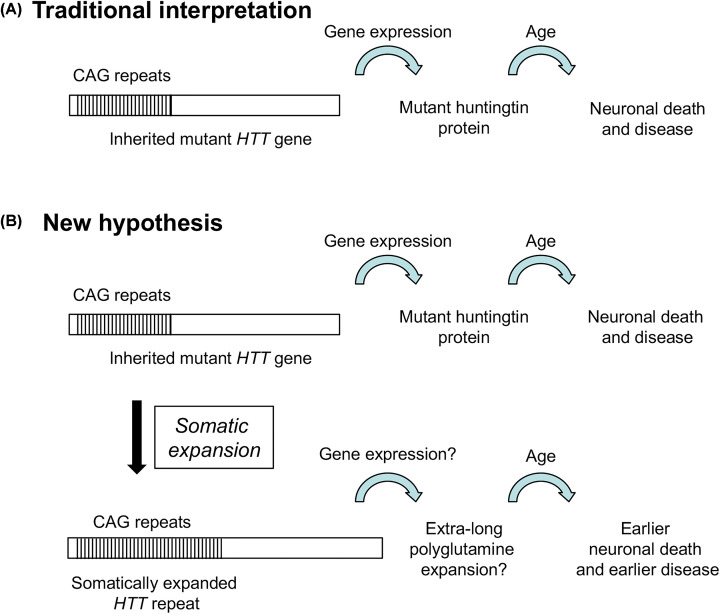
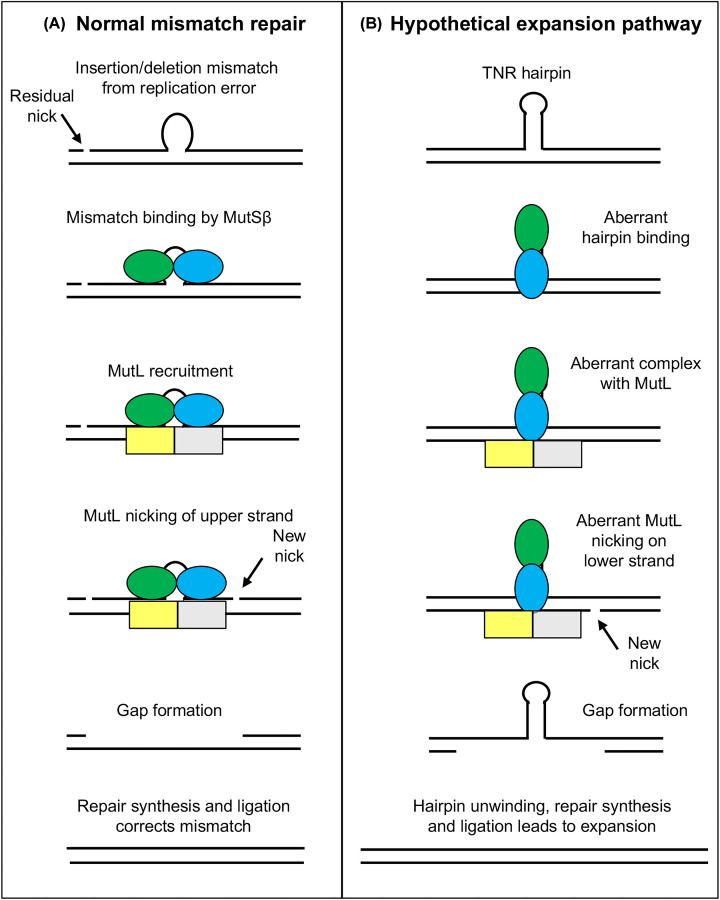
Huntington's disease (HD) is a fatal, inherited neurodegenerative disease that causes neuronal death, particularly in medium spiny neurons. HD leads to serious and progressive motor, cognitive and psychiatric symptoms. Its genetic basis is an expansion of the CAG triplet repeat in the HTT gene, leading to extra glutamines in the huntingtin protein. HD is one of nine genetic diseases in this polyglutamine (polyQ) category, that also includes a number of inherited spinocerebellar ataxias (SCAs). Traditionally it has been assumed that HD age of onset and disease progression were solely the outcome of age-dependent exposure of neurons to toxic effects of the inherited mutant huntingtin protein. However, recent genome-wide association studies (GWAS) have revealed significant effects of genetic variants outside of HTT. Surprisingly, these variants turn out to be mostly in genes encoding DNA repair factors, suggesting that at least some disease modulation occurs at the level of the HTT DNA itself. These DNA repair proteins are known from model systems to promote ongoing somatic CAG repeat expansions in tissues affected by HD. Thus, for triplet repeats, some DNA repair proteins seem to abandon their normal genoprotective roles and, instead, drive expansions and accelerate disease. One attractive hypothesis-still to be proven rigorously-is that somatic HTT expansions augment the disease burden of the inherited allele. If so, therapeutic approaches that lower levels of huntingtin protein may need blending with additional therapies that reduce levels of somatic CAG repeat expansions to achieve maximal effect.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
