{"title":"鉴定一种新的MICU1无义变异导致伊朗近亲家族锥体外系症状的肌病。","authors":"Fatemeh Bitarafan, Mehrnoosh Khodaeian, Elham Amjadi Sardehaei, Fatemeh Zahra Darvishi, Navid Almadani, Yalda Nilipour, Masoud Garshasbi","doi":"10.1186/s40348-021-00116-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Ca<sup>2+</sup> as a universal second messenger regulates basic biological functions including cell cycle, cell proliferation, cell differentiation, and cell death. Lack of the protein mitochondrial calcium uptake1 (MICU1), which has been regarded as a gatekeeper of Ca ions, leads to the abnormal mitochondrial Ca<sup>2+</sup> handling, excessive production of reactive oxygen species (ROS), and increased cell death. Mutations in MICU1 gene causes a very rare neuromuscular disease, myopathy with extrapyramidal signs (MPXPS), due to primary alterations in mitochondrial calcium signaling which demonstrates the key role of mitochondrial Ca<sup>2+</sup> uptake. To date, 13 variants have been reported in MICU1 gene in 44 patients presented with the vast spectrum of symptoms.</p><p><strong>Case presentation: </strong>Here, we report a 44-year-old Iranian patient presented with learning disability, muscle weakness, easy fatigability, reduced tendon reflexes, ataxia, gait disturbance, elevated hepatic transaminases, elevated serum creatine kinase (CK), and elevated lactate dehydrogenase (LDH). We identified a novel nonsense variant c.385C>T; p.(R129*) in MICU1 gene by whole exome sequencing (WES) and segregation analysis.</p><p><strong>Conclusions: </strong>Our finding along with previous studies provides more evidence on the clinical presentation of the disease caused by pathogenic mutations in MICU1. Finding more variants and expanding the spectrum of the disease increases the diagnostic rate of molecular testing in screening of this kind of diseases and in turn improves the quality of counseling for at risk couples and helps them to minimize the risks of having affected children.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"8 1","pages":"6"},"PeriodicalIF":3.4000,"publicationDate":"2021-05-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8107061/pdf/","citationCount":"7","resultStr":"{\"title\":\"Identification of a novel MICU1 nonsense variant causes myopathy with extrapyramidal signs in an Iranian consanguineous family.\",\"authors\":\"Fatemeh Bitarafan, Mehrnoosh Khodaeian, Elham Amjadi Sardehaei, Fatemeh Zahra Darvishi, Navid Almadani, Yalda Nilipour, Masoud Garshasbi\",\"doi\":\"10.1186/s40348-021-00116-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Ca<sup>2+</sup> as a universal second messenger regulates basic biological functions including cell cycle, cell proliferation, cell differentiation, and cell death. Lack of the protein mitochondrial calcium uptake1 (MICU1), which has been regarded as a gatekeeper of Ca ions, leads to the abnormal mitochondrial Ca<sup>2+</sup> handling, excessive production of reactive oxygen species (ROS), and increased cell death. Mutations in MICU1 gene causes a very rare neuromuscular disease, myopathy with extrapyramidal signs (MPXPS), due to primary alterations in mitochondrial calcium signaling which demonstrates the key role of mitochondrial Ca<sup>2+</sup> uptake. To date, 13 variants have been reported in MICU1 gene in 44 patients presented with the vast spectrum of symptoms.</p><p><strong>Case presentation: </strong>Here, we report a 44-year-old Iranian patient presented with learning disability, muscle weakness, easy fatigability, reduced tendon reflexes, ataxia, gait disturbance, elevated hepatic transaminases, elevated serum creatine kinase (CK), and elevated lactate dehydrogenase (LDH). We identified a novel nonsense variant c.385C>T; p.(R129*) in MICU1 gene by whole exome sequencing (WES) and segregation analysis.</p><p><strong>Conclusions: </strong>Our finding along with previous studies provides more evidence on the clinical presentation of the disease caused by pathogenic mutations in MICU1. Finding more variants and expanding the spectrum of the disease increases the diagnostic rate of molecular testing in screening of this kind of diseases and in turn improves the quality of counseling for at risk couples and helps them to minimize the risks of having affected children.</p>\",\"PeriodicalId\":74215,\"journal\":{\"name\":\"Molecular and cellular pediatrics\",\"volume\":\"8 1\",\"pages\":\"6\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2021-05-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8107061/pdf/\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular and cellular pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40348-021-00116-w\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-021-00116-w","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Identification of a novel MICU1 nonsense variant causes myopathy with extrapyramidal signs in an Iranian consanguineous family.
Background: Ca2+ as a universal second messenger regulates basic biological functions including cell cycle, cell proliferation, cell differentiation, and cell death. Lack of the protein mitochondrial calcium uptake1 (MICU1), which has been regarded as a gatekeeper of Ca ions, leads to the abnormal mitochondrial Ca2+ handling, excessive production of reactive oxygen species (ROS), and increased cell death. Mutations in MICU1 gene causes a very rare neuromuscular disease, myopathy with extrapyramidal signs (MPXPS), due to primary alterations in mitochondrial calcium signaling which demonstrates the key role of mitochondrial Ca2+ uptake. To date, 13 variants have been reported in MICU1 gene in 44 patients presented with the vast spectrum of symptoms.
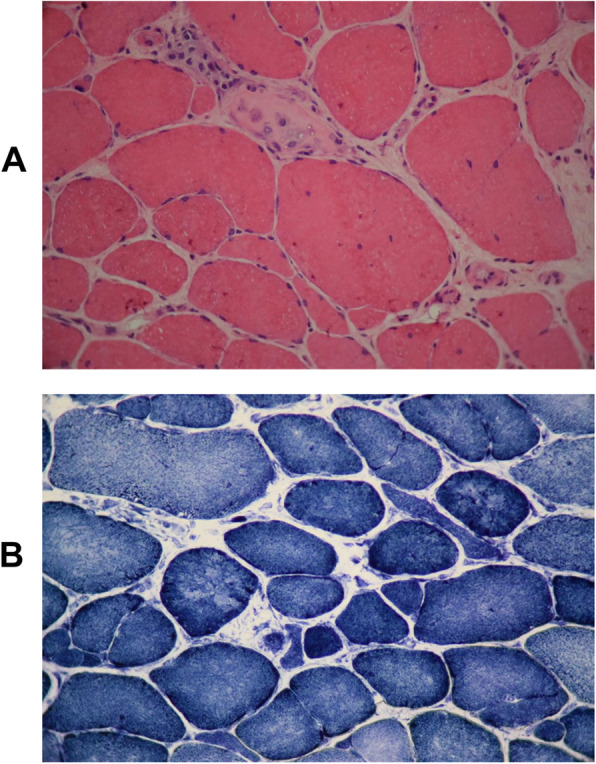
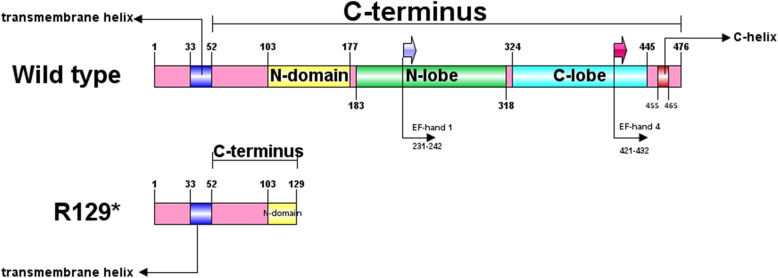
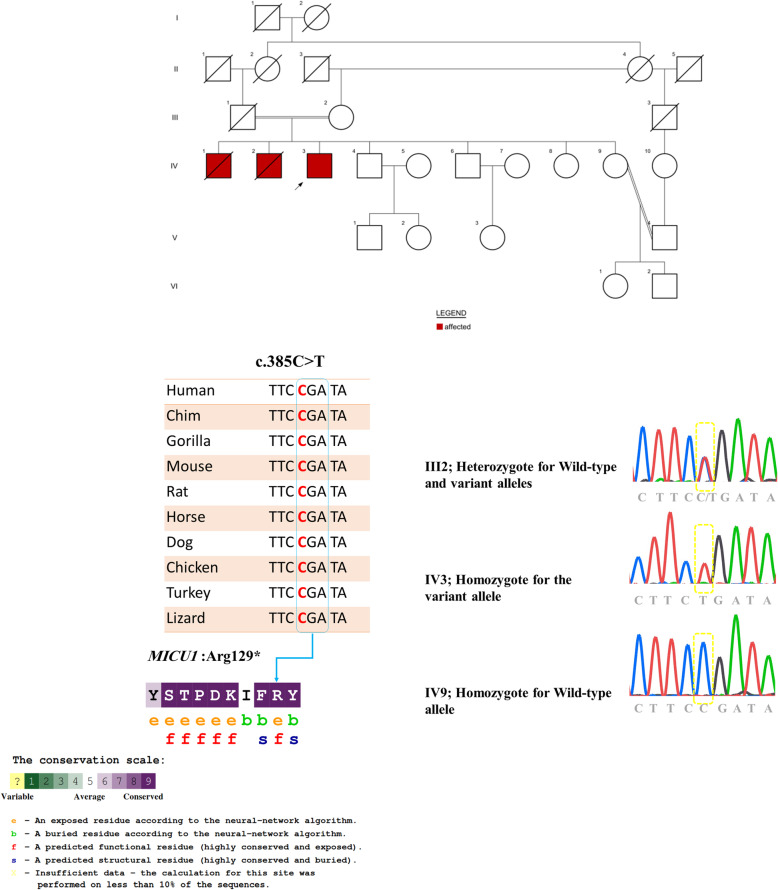
Case presentation: Here, we report a 44-year-old Iranian patient presented with learning disability, muscle weakness, easy fatigability, reduced tendon reflexes, ataxia, gait disturbance, elevated hepatic transaminases, elevated serum creatine kinase (CK), and elevated lactate dehydrogenase (LDH). We identified a novel nonsense variant c.385C>T; p.(R129*) in MICU1 gene by whole exome sequencing (WES) and segregation analysis.
Conclusions: Our finding along with previous studies provides more evidence on the clinical presentation of the disease caused by pathogenic mutations in MICU1. Finding more variants and expanding the spectrum of the disease increases the diagnostic rate of molecular testing in screening of this kind of diseases and in turn improves the quality of counseling for at risk couples and helps them to minimize the risks of having affected children.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
