Khushnooda Ramzan, Maha Alotaibi, Rozeena Huma, Sibtain Afzal
{"title":"与隐性成骨不全相关的复发性TMEM38B基因缺失的检测。","authors":"Khushnooda Ramzan, Maha Alotaibi, Rozeena Huma, Sibtain Afzal","doi":"10.15190/d.2021.3","DOIUrl":null,"url":null,"abstract":"<p><p>Osteogenesis imperfecta is a clinically and genetically group of heterogeneous disorders associated with decreased bone density, brittle bones, bone deformity, recurrent fractures, and growth retardation. Osteogenesis imperfecta is commonly associated with mutations of the genes encoding for type I collagen (COL1A1/COL1A2). Mutations in other genes, some associated with type I collagen post-translational processing, have also been identified as the cause of osteogenesis imperfecta. Mutations in the transmembrane protein 38B (TMEM38B) gene have been reported in a rare autosomal recessive form of osteogenesis imperfecta. TMEM38B encodes TRIC-B - a trimeric intracellular cation channel type B which is essential to modulate intracellular calcium signaling. In this study, we are reporting a case of osteogenesis imperfecta type XIV from a Saudi consanguineous family. Our patient was an eight-month-old child with short limbs, club feet, and lower limb deformities with developmental delay. Radiological findings were consistent with the evidence of osteogenesis imperfecta. There was no evidence of impaired hearing or blue sclera and based on the clinical assessment, we classified our patient as a non-syndromic osteogenesis imperfecta. A pathogenic deletion in the chromosome 9q31.2 region, partially encompassing the TMEM38B gene, was detected using chromosomal microarray analysis. This study expands our knowledge about the rare type of osteogenesis imperfecta in our consanguineous population. Besides, it emphasizes the use of genomic medicine in clinical practices to formulate early interventions to clinically improve the patient's condition.</p>","PeriodicalId":72829,"journal":{"name":"Discoveries (Craiova, Romania)","volume":"9 1","pages":"e124"},"PeriodicalIF":0.0000,"publicationDate":"2021-03-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8140756/pdf/","citationCount":"3","resultStr":"{\"title\":\"Detection of a Recurrent TMEM38B Gene Deletion Associated with Recessive Osteogenesis Imperfecta.\",\"authors\":\"Khushnooda Ramzan, Maha Alotaibi, Rozeena Huma, Sibtain Afzal\",\"doi\":\"10.15190/d.2021.3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Osteogenesis imperfecta is a clinically and genetically group of heterogeneous disorders associated with decreased bone density, brittle bones, bone deformity, recurrent fractures, and growth retardation. Osteogenesis imperfecta is commonly associated with mutations of the genes encoding for type I collagen (COL1A1/COL1A2). Mutations in other genes, some associated with type I collagen post-translational processing, have also been identified as the cause of osteogenesis imperfecta. Mutations in the transmembrane protein 38B (TMEM38B) gene have been reported in a rare autosomal recessive form of osteogenesis imperfecta. TMEM38B encodes TRIC-B - a trimeric intracellular cation channel type B which is essential to modulate intracellular calcium signaling. In this study, we are reporting a case of osteogenesis imperfecta type XIV from a Saudi consanguineous family. Our patient was an eight-month-old child with short limbs, club feet, and lower limb deformities with developmental delay. Radiological findings were consistent with the evidence of osteogenesis imperfecta. There was no evidence of impaired hearing or blue sclera and based on the clinical assessment, we classified our patient as a non-syndromic osteogenesis imperfecta. A pathogenic deletion in the chromosome 9q31.2 region, partially encompassing the TMEM38B gene, was detected using chromosomal microarray analysis. This study expands our knowledge about the rare type of osteogenesis imperfecta in our consanguineous population. Besides, it emphasizes the use of genomic medicine in clinical practices to formulate early interventions to clinically improve the patient's condition.</p>\",\"PeriodicalId\":72829,\"journal\":{\"name\":\"Discoveries (Craiova, Romania)\",\"volume\":\"9 1\",\"pages\":\"e124\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-03-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8140756/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Discoveries (Craiova, Romania)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.15190/d.2021.3\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Discoveries (Craiova, Romania)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.15190/d.2021.3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Detection of a Recurrent TMEM38B Gene Deletion Associated with Recessive Osteogenesis Imperfecta.
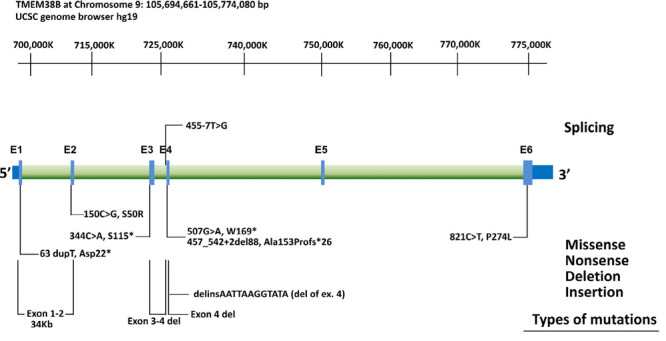
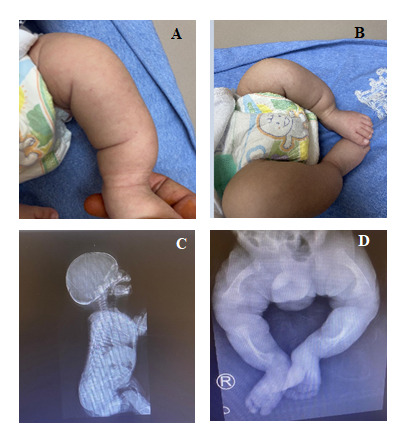
Osteogenesis imperfecta is a clinically and genetically group of heterogeneous disorders associated with decreased bone density, brittle bones, bone deformity, recurrent fractures, and growth retardation. Osteogenesis imperfecta is commonly associated with mutations of the genes encoding for type I collagen (COL1A1/COL1A2). Mutations in other genes, some associated with type I collagen post-translational processing, have also been identified as the cause of osteogenesis imperfecta. Mutations in the transmembrane protein 38B (TMEM38B) gene have been reported in a rare autosomal recessive form of osteogenesis imperfecta. TMEM38B encodes TRIC-B - a trimeric intracellular cation channel type B which is essential to modulate intracellular calcium signaling. In this study, we are reporting a case of osteogenesis imperfecta type XIV from a Saudi consanguineous family. Our patient was an eight-month-old child with short limbs, club feet, and lower limb deformities with developmental delay. Radiological findings were consistent with the evidence of osteogenesis imperfecta. There was no evidence of impaired hearing or blue sclera and based on the clinical assessment, we classified our patient as a non-syndromic osteogenesis imperfecta. A pathogenic deletion in the chromosome 9q31.2 region, partially encompassing the TMEM38B gene, was detected using chromosomal microarray analysis. This study expands our knowledge about the rare type of osteogenesis imperfecta in our consanguineous population. Besides, it emphasizes the use of genomic medicine in clinical practices to formulate early interventions to clinically improve the patient's condition.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
