Prashant Sharma , Elena-Raluca Nicoli , Jenny Serra-Vinardell , Marie Morimoto , Camilo Toro , May Christine V. Malicdan , Wendy J. Introne
{"title":"Chediak-Higashi综合征:回顾过去,现在和未来","authors":"Prashant Sharma , Elena-Raluca Nicoli , Jenny Serra-Vinardell , Marie Morimoto , Camilo Toro , May Christine V. Malicdan , Wendy J. Introne","doi":"10.1016/j.ddmod.2019.10.008","DOIUrl":null,"url":null,"abstract":"<div><p><span>Since the initial description of Chediak-Higashi syndrome (CHS), over 75 years ago, several studies have been conducted to underscore the role of the lysosomal trafficking regulator (</span><em>LYST</em><span>) gene in the pathogenesis of disease. CHS is a rare autosomal recessive disorder, which is caused by biallelic mutations in the highly conserved </span><em>LYST</em><span><span> gene. The disease is characterized by partial oculocutaneous albinism<span>, prolonged bleeding, immune and neurologic dysfunction, and risk for the development of hemophagocytic lympohistiocytosis (HLH). The presence of giant secretory granules in leukocytes is the classical diagnostic feature, which distinguishes CHS from closely related Griscelli and Hermansky-Pudlak syndromes. While the exact mechanism of the formation of the giant granules in CHS patients is not understood, dysregulation of LYST function in regulating lysosomal biogenesis has been proposed to play a role. In this review, we discuss the clinical characteristics of the disease and highlight the functional consequences of enlarged </span></span>lysosomes and lysosome-related organelles (LROs) in CHS.</span></p></div>","PeriodicalId":39774,"journal":{"name":"Drug Discovery Today: Disease Models","volume":"31 ","pages":"Pages 31-36"},"PeriodicalIF":0.0000,"publicationDate":"2020-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.ddmod.2019.10.008","citationCount":"23","resultStr":"{\"title\":\"Chediak-Higashi syndrome: A review of the past, present, and future\",\"authors\":\"Prashant Sharma , Elena-Raluca Nicoli , Jenny Serra-Vinardell , Marie Morimoto , Camilo Toro , May Christine V. Malicdan , Wendy J. Introne\",\"doi\":\"10.1016/j.ddmod.2019.10.008\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><span>Since the initial description of Chediak-Higashi syndrome (CHS), over 75 years ago, several studies have been conducted to underscore the role of the lysosomal trafficking regulator (</span><em>LYST</em><span>) gene in the pathogenesis of disease. CHS is a rare autosomal recessive disorder, which is caused by biallelic mutations in the highly conserved </span><em>LYST</em><span><span> gene. The disease is characterized by partial oculocutaneous albinism<span>, prolonged bleeding, immune and neurologic dysfunction, and risk for the development of hemophagocytic lympohistiocytosis (HLH). The presence of giant secretory granules in leukocytes is the classical diagnostic feature, which distinguishes CHS from closely related Griscelli and Hermansky-Pudlak syndromes. While the exact mechanism of the formation of the giant granules in CHS patients is not understood, dysregulation of LYST function in regulating lysosomal biogenesis has been proposed to play a role. In this review, we discuss the clinical characteristics of the disease and highlight the functional consequences of enlarged </span></span>lysosomes and lysosome-related organelles (LROs) in CHS.</span></p></div>\",\"PeriodicalId\":39774,\"journal\":{\"name\":\"Drug Discovery Today: Disease Models\",\"volume\":\"31 \",\"pages\":\"Pages 31-36\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.ddmod.2019.10.008\",\"citationCount\":\"23\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Drug Discovery Today: Disease Models\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1740675719300386\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/12/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"Pharmacology, Toxicology and Pharmaceutics\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Drug Discovery Today: Disease Models","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1740675719300386","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/12/9 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Pharmacology, Toxicology and Pharmaceutics","Score":null,"Total":0}
Chediak-Higashi syndrome: A review of the past, present, and future
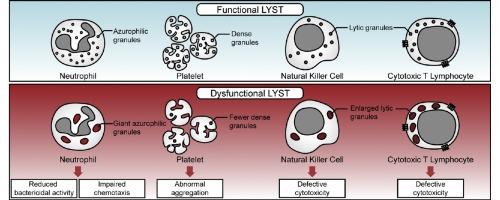
Since the initial description of Chediak-Higashi syndrome (CHS), over 75 years ago, several studies have been conducted to underscore the role of the lysosomal trafficking regulator (LYST) gene in the pathogenesis of disease. CHS is a rare autosomal recessive disorder, which is caused by biallelic mutations in the highly conserved LYST gene. The disease is characterized by partial oculocutaneous albinism, prolonged bleeding, immune and neurologic dysfunction, and risk for the development of hemophagocytic lympohistiocytosis (HLH). The presence of giant secretory granules in leukocytes is the classical diagnostic feature, which distinguishes CHS from closely related Griscelli and Hermansky-Pudlak syndromes. While the exact mechanism of the formation of the giant granules in CHS patients is not understood, dysregulation of LYST function in regulating lysosomal biogenesis has been proposed to play a role. In this review, we discuss the clinical characteristics of the disease and highlight the functional consequences of enlarged lysosomes and lysosome-related organelles (LROs) in CHS.
期刊介绍:
Drug Discovery Today: Disease Models discusses the non-human experimental models through which inference is drawn regarding the molecular aetiology and pathogenesis of human disease. It provides critical analysis and evaluation of which models can genuinely inform the research community about the direct process of human disease, those which may have value in basic toxicology, and those which are simply designed for effective expression and raw characterisation.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
