Krithika Murali, Tanya M Dwarte, Mehrdad Nikfarjam, Katherine M Tucker, Rhys B Vaughan, Marios Efthymiou, Allison Collins, Allan D Spigelman, Lucinda Salmon, Amber L Johns, David B Williams, Martin B Delatycki, Thomas John, Alina Stoita
{"title":"澳大利亚胰腺癌筛查项目参与者中新的生殖系致病变异的显著检测。","authors":"Krithika Murali, Tanya M Dwarte, Mehrdad Nikfarjam, Katherine M Tucker, Rhys B Vaughan, Marios Efthymiou, Allison Collins, Allan D Spigelman, Lucinda Salmon, Amber L Johns, David B Williams, Martin B Delatycki, Thomas John, Alina Stoita","doi":"10.1186/s13053-021-00190-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The Australian Pancreatic Cancer Screening Program (APCSP) offers endoscopic ultrasound surveillance for individuals at increased risk of pancreatic ductal adenocarcinoma (PDAC) with all participants requiring assessment by a Familial Cancer Service before or after study enrolment.</p><p><strong>Methods: </strong>Individuals aged 40-80 years (or 10 years younger than the earliest PDAC diagnosis) were eligible for APCSP study entry if they had 1) ≥ two blood relatives with PDAC (at least one of first-degree association); 2) a clinical or genetic diagnosis of Hereditary Pancreatitis or Peutz-Jeghers syndrome irrespective of PDAC family history; or 3) a known PDAC predisposition germline pathogenic variant (BRCA2, PALB2, CDKN2A, or Lynch syndrome) with ≥one PDAC-affected first- or second-degree relative. Retrospective medical record review was conducted for APCSP participants enrolled at the participating Australian hospitals from January 2011 to December 2019. We audited the genetic investigations offered by multiple Familial Cancer Services who assessed APCSP participants according to national guidelines, local clinical protocol and/or the availability of external research-funded testing, and the subsequent findings. Descriptive statistical analysis was performed using Microsoft Excel.</p><p><strong>Results: </strong>Of 189 kindreds (285 participants), 50 kindreds (71 participants) had a known germline pathogenic variant at enrolment (BRCA2 n = 35, PALB2 n = 6, CDKN2A n = 3, STK11 n = 3, PRSS1 n = 2, MLH1 n = 1). Forty-eight of 136 (35%) kindreds with no known germline pathogenic variant were offered mutation analysis; 89% was clinic-funded, with increasing self-funded testing since 2016. The relatively low rates of genetic testing performed reflects initial strict criteria for clinic-funded genetic testing. New germline pathogenic variants were detected in five kindreds (10.4%) after study enrolment (BRCA2 n = 3 kindreds, PALB2 n = 1, CDKN2A n = 1). Of note, only eight kindreds were reassessed by a Familial Cancer Service since enrolment, with a further 21 kindreds identified as being suitable for reassessment.</p><p><strong>Conclusion: </strong>Germline pathogenic variants associated with PDAC were seen in 29.1% of our high-risk cohort (55/189 kindreds; 82/285 participants). Importantly, 10.4% of kindreds offered genetic testing were newly identified as having germline pathogenic variants, with majority being BRCA2. As genetic testing standards evolve rapidly in PDAC, 5-yearly reassessment of high-risk individuals by Familial Cancer Services is warranted.</p>","PeriodicalId":55058,"journal":{"name":"Hereditary Cancer in Clinical Practice","volume":"19 1","pages":"33"},"PeriodicalIF":2.4000,"publicationDate":"2021-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8365963/pdf/","citationCount":"2","resultStr":"{\"title\":\"Significant detection of new germline pathogenic variants in Australian Pancreatic Cancer Screening Program participants.\",\"authors\":\"Krithika Murali, Tanya M Dwarte, Mehrdad Nikfarjam, Katherine M Tucker, Rhys B Vaughan, Marios Efthymiou, Allison Collins, Allan D Spigelman, Lucinda Salmon, Amber L Johns, David B Williams, Martin B Delatycki, Thomas John, Alina Stoita\",\"doi\":\"10.1186/s13053-021-00190-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The Australian Pancreatic Cancer Screening Program (APCSP) offers endoscopic ultrasound surveillance for individuals at increased risk of pancreatic ductal adenocarcinoma (PDAC) with all participants requiring assessment by a Familial Cancer Service before or after study enrolment.</p><p><strong>Methods: </strong>Individuals aged 40-80 years (or 10 years younger than the earliest PDAC diagnosis) were eligible for APCSP study entry if they had 1) ≥ two blood relatives with PDAC (at least one of first-degree association); 2) a clinical or genetic diagnosis of Hereditary Pancreatitis or Peutz-Jeghers syndrome irrespective of PDAC family history; or 3) a known PDAC predisposition germline pathogenic variant (BRCA2, PALB2, CDKN2A, or Lynch syndrome) with ≥one PDAC-affected first- or second-degree relative. Retrospective medical record review was conducted for APCSP participants enrolled at the participating Australian hospitals from January 2011 to December 2019. We audited the genetic investigations offered by multiple Familial Cancer Services who assessed APCSP participants according to national guidelines, local clinical protocol and/or the availability of external research-funded testing, and the subsequent findings. Descriptive statistical analysis was performed using Microsoft Excel.</p><p><strong>Results: </strong>Of 189 kindreds (285 participants), 50 kindreds (71 participants) had a known germline pathogenic variant at enrolment (BRCA2 n = 35, PALB2 n = 6, CDKN2A n = 3, STK11 n = 3, PRSS1 n = 2, MLH1 n = 1). Forty-eight of 136 (35%) kindreds with no known germline pathogenic variant were offered mutation analysis; 89% was clinic-funded, with increasing self-funded testing since 2016. The relatively low rates of genetic testing performed reflects initial strict criteria for clinic-funded genetic testing. New germline pathogenic variants were detected in five kindreds (10.4%) after study enrolment (BRCA2 n = 3 kindreds, PALB2 n = 1, CDKN2A n = 1). Of note, only eight kindreds were reassessed by a Familial Cancer Service since enrolment, with a further 21 kindreds identified as being suitable for reassessment.</p><p><strong>Conclusion: </strong>Germline pathogenic variants associated with PDAC were seen in 29.1% of our high-risk cohort (55/189 kindreds; 82/285 participants). Importantly, 10.4% of kindreds offered genetic testing were newly identified as having germline pathogenic variants, with majority being BRCA2. As genetic testing standards evolve rapidly in PDAC, 5-yearly reassessment of high-risk individuals by Familial Cancer Services is warranted.</p>\",\"PeriodicalId\":55058,\"journal\":{\"name\":\"Hereditary Cancer in Clinical Practice\",\"volume\":\"19 1\",\"pages\":\"33\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2021-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8365963/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hereditary Cancer in Clinical Practice\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13053-021-00190-1\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditary Cancer in Clinical Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13053-021-00190-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 2
摘要
背景:澳大利亚癌症筛查计划(APCSP)为胰腺导管腺癌(PDAC)风险增加的个体提供内窥镜超声监测,所有参与者在研究注册前或注册后都需要家族癌症服务机构的评估。方法:40-80岁的个体 年(或10 比最早的PDAC诊断年轻几岁)有资格参加APCSP研究,如果他们有1) ≥ 两名患有PDAC的血亲(至少一名为一级关联);2) 遗传性胰腺炎或Peutz-Jeghers综合征的临床或遗传诊断,与PDAC家族史无关;或3)已知的PDAC易感性种系致病性变体(BRCA2、PALB2、CDKN2A或Lynch综合征),具有≥一个受PDAC影响的一级或二级亲属。对2011年1月至2019年12月在参与的澳大利亚医院登记的APCSP参与者进行了回顾性医疗记录审查。我们审计了多个癌症家族服务机构提供的基因调查,这些服务机构根据国家指南、当地临床方案和/或外部研究资助测试的可用性以及随后的发现对APCSP参与者进行了评估。结果:在189个家系(285名参与者)中,50个家系在登记时有已知的种系致病性变异(BRCA2 n = 35,PALB2 n = 6,CDKN2A n = 3,STK11 n = 3,PRSS1 n = 2,MLH1 n = 1) 。136个没有已知种系致病变异株的家系中有48个(35%)进行了突变分析;89%由诊所资助,自2016年以来,自费检测不断增加。进行基因检测的比率相对较低,这反映了临床资助的基因检测最初的严格标准。研究登记后,在5个家系(10.4%)中检测到新的种系致病性变异(BRCA2 n = 3个家族,PALB2 n = 1,CDKN2A n = 1) 。值得注意的是,自注册以来,癌症家族服务机构仅对8个品种进行了重新评估,另有21个品种被确定为适合重新评估。结论:29.1%的高危队列(55/189个家系;82/285名参与者)中发现了与PDAC相关的种系致病性变异。重要的是,10.4%的提供基因检测的家族被新发现具有种系致病性变体,其中大多数是BRCA2。随着PDAC基因检测标准的快速发展,癌症家族服务机构有必要对高危人群进行5年一次的重新评估。
Significant detection of new germline pathogenic variants in Australian Pancreatic Cancer Screening Program participants.
Background: The Australian Pancreatic Cancer Screening Program (APCSP) offers endoscopic ultrasound surveillance for individuals at increased risk of pancreatic ductal adenocarcinoma (PDAC) with all participants requiring assessment by a Familial Cancer Service before or after study enrolment.
Methods: Individuals aged 40-80 years (or 10 years younger than the earliest PDAC diagnosis) were eligible for APCSP study entry if they had 1) ≥ two blood relatives with PDAC (at least one of first-degree association); 2) a clinical or genetic diagnosis of Hereditary Pancreatitis or Peutz-Jeghers syndrome irrespective of PDAC family history; or 3) a known PDAC predisposition germline pathogenic variant (BRCA2, PALB2, CDKN2A, or Lynch syndrome) with ≥one PDAC-affected first- or second-degree relative. Retrospective medical record review was conducted for APCSP participants enrolled at the participating Australian hospitals from January 2011 to December 2019. We audited the genetic investigations offered by multiple Familial Cancer Services who assessed APCSP participants according to national guidelines, local clinical protocol and/or the availability of external research-funded testing, and the subsequent findings. Descriptive statistical analysis was performed using Microsoft Excel.
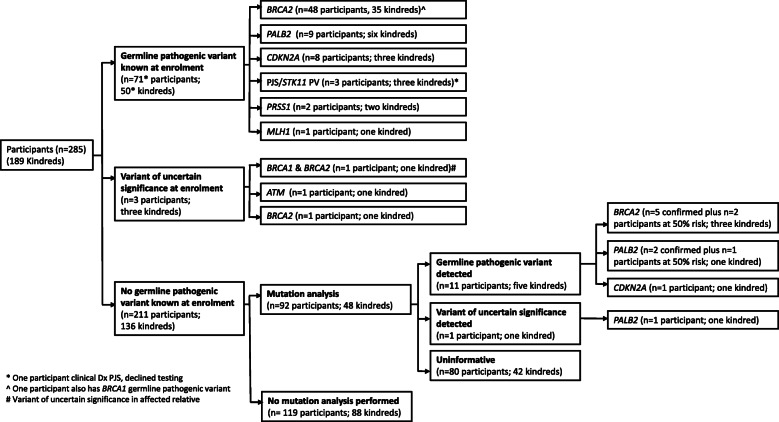
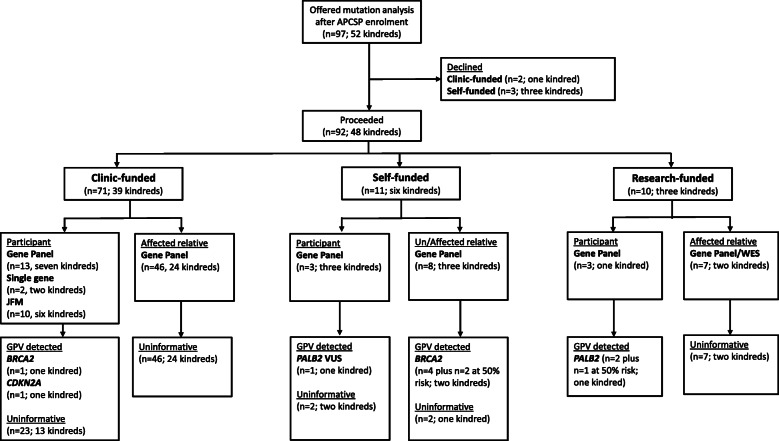
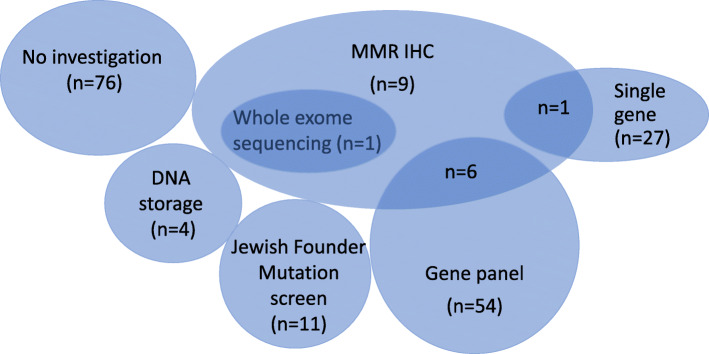
Results: Of 189 kindreds (285 participants), 50 kindreds (71 participants) had a known germline pathogenic variant at enrolment (BRCA2 n = 35, PALB2 n = 6, CDKN2A n = 3, STK11 n = 3, PRSS1 n = 2, MLH1 n = 1). Forty-eight of 136 (35%) kindreds with no known germline pathogenic variant were offered mutation analysis; 89% was clinic-funded, with increasing self-funded testing since 2016. The relatively low rates of genetic testing performed reflects initial strict criteria for clinic-funded genetic testing. New germline pathogenic variants were detected in five kindreds (10.4%) after study enrolment (BRCA2 n = 3 kindreds, PALB2 n = 1, CDKN2A n = 1). Of note, only eight kindreds were reassessed by a Familial Cancer Service since enrolment, with a further 21 kindreds identified as being suitable for reassessment.
Conclusion: Germline pathogenic variants associated with PDAC were seen in 29.1% of our high-risk cohort (55/189 kindreds; 82/285 participants). Importantly, 10.4% of kindreds offered genetic testing were newly identified as having germline pathogenic variants, with majority being BRCA2. As genetic testing standards evolve rapidly in PDAC, 5-yearly reassessment of high-risk individuals by Familial Cancer Services is warranted.
期刊介绍:
Hereditary Cancer in Clinical Practice is an open access journal that publishes articles of interest for the cancer genetics community and serves as a discussion forum for the development appropriate healthcare strategies.
Cancer genetics encompasses a wide variety of disciplines and knowledge in the field is rapidly growing, especially as the amount of information linking genetic differences to inherited cancer predispositions continues expanding. With the increased knowledge of genetic variability and how this relates to cancer risk there is a growing demand not only to disseminate this information into clinical practice but also to enable competent debate concerning how such information is managed and what it implies for patient care.
Topics covered by the journal include but are not limited to:
Original research articles on any aspect of inherited predispositions to cancer.
Reviews of inherited cancer predispositions.
Application of molecular and cytogenetic analysis to clinical decision making.
Clinical aspects of the management of hereditary cancers.
Genetic counselling issues associated with cancer genetics.
The role of registries in improving health care of patients with an inherited predisposition to cancer.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
