{"title":"轻、重度钩端螺旋体患者钩端螺旋体基因组比较分析。","authors":"Songtham Anuntakarun, Vorthon Sawaswong, Rungrat Jitvaropas, Kesmanee Praianantathavorn, Witthaya Poomipak, Yupin Suputtamongkol, Chintana Chirathaworn, Sunchai Payungporn","doi":"10.5808/gi.21037","DOIUrl":null,"url":null,"abstract":"<p><p>Leptospirosis is a zoonotic disease caused by spirochetes from the genus Leptospira. In Thailand, Leptospira interrogans is a major cause of leptospirosis. Leptospirosis patients present with a wide range of clinical manifestations from asymptomatic, mild infections to severe illness involving organ failure. For better understanding the difference between Leptospira isolates causing mild and severe leptospirosis, illumina sequencing was used to sequence genomic DNA in both serotypes. DNA of Leptospira isolated from two patients, one with mild and another with severe symptoms, were included in this study. The paired-end reads were removed adapters and trimmed with Q30 score using Trimmomatic. Trimmed reads were constructed to contigs and scaffolds using SPAdes. Cross-contamination of scaffolds was evaluated by ContEst16s. Prokka tool for bacterial annotation was used to annotate sequences from both Leptospira isolates. Predicted amino acid sequences from Prokka were searched in EggNOG and David gene ontology database to characterize gene ontology. In addition, Leptospira from mild and severe patients, that passed the criteria e-value < 10e-5 from blastP against virulence factor database, were used to analyze with Venn diagram. From this study, we found 13 and 12 genes that were unique in the isolates from mild and severe patients, respectively. The 12 genes in the severe isolate might be virulence factor genes that affect disease severity. However, these genes should be validated in further study.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"19 3","pages":"e31"},"PeriodicalIF":0.0000,"publicationDate":"2021-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8510873/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparative genome characterization of Leptospira interrogans from mild and severe leptospirosis patients.\",\"authors\":\"Songtham Anuntakarun, Vorthon Sawaswong, Rungrat Jitvaropas, Kesmanee Praianantathavorn, Witthaya Poomipak, Yupin Suputtamongkol, Chintana Chirathaworn, Sunchai Payungporn\",\"doi\":\"10.5808/gi.21037\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Leptospirosis is a zoonotic disease caused by spirochetes from the genus Leptospira. In Thailand, Leptospira interrogans is a major cause of leptospirosis. Leptospirosis patients present with a wide range of clinical manifestations from asymptomatic, mild infections to severe illness involving organ failure. For better understanding the difference between Leptospira isolates causing mild and severe leptospirosis, illumina sequencing was used to sequence genomic DNA in both serotypes. DNA of Leptospira isolated from two patients, one with mild and another with severe symptoms, were included in this study. The paired-end reads were removed adapters and trimmed with Q30 score using Trimmomatic. Trimmed reads were constructed to contigs and scaffolds using SPAdes. Cross-contamination of scaffolds was evaluated by ContEst16s. Prokka tool for bacterial annotation was used to annotate sequences from both Leptospira isolates. Predicted amino acid sequences from Prokka were searched in EggNOG and David gene ontology database to characterize gene ontology. In addition, Leptospira from mild and severe patients, that passed the criteria e-value < 10e-5 from blastP against virulence factor database, were used to analyze with Venn diagram. From this study, we found 13 and 12 genes that were unique in the isolates from mild and severe patients, respectively. The 12 genes in the severe isolate might be virulence factor genes that affect disease severity. However, these genes should be validated in further study.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\"19 3\",\"pages\":\"e31\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8510873/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.21037\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/9/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.21037","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/9/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Comparative genome characterization of Leptospira interrogans from mild and severe leptospirosis patients.
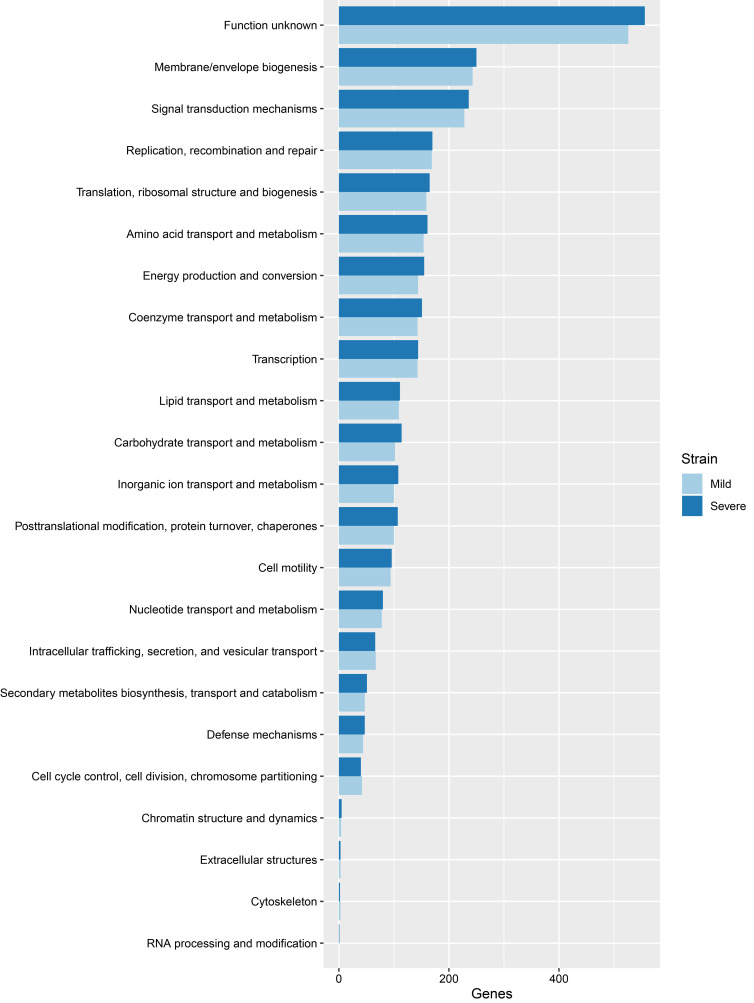
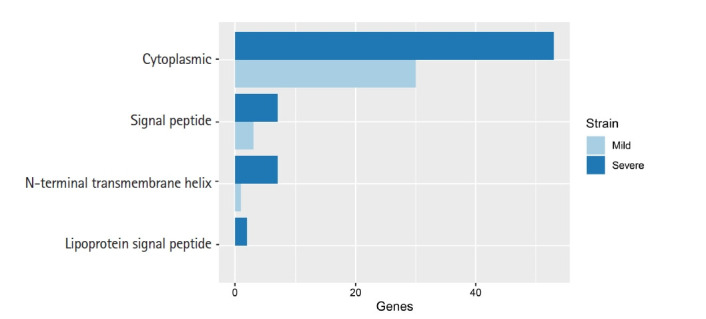
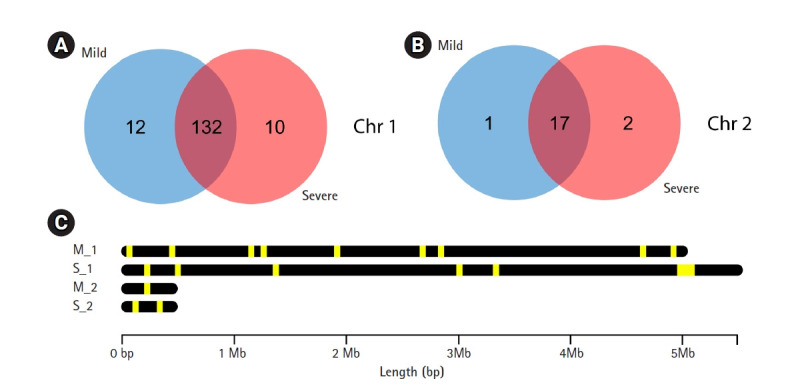
Leptospirosis is a zoonotic disease caused by spirochetes from the genus Leptospira. In Thailand, Leptospira interrogans is a major cause of leptospirosis. Leptospirosis patients present with a wide range of clinical manifestations from asymptomatic, mild infections to severe illness involving organ failure. For better understanding the difference between Leptospira isolates causing mild and severe leptospirosis, illumina sequencing was used to sequence genomic DNA in both serotypes. DNA of Leptospira isolated from two patients, one with mild and another with severe symptoms, were included in this study. The paired-end reads were removed adapters and trimmed with Q30 score using Trimmomatic. Trimmed reads were constructed to contigs and scaffolds using SPAdes. Cross-contamination of scaffolds was evaluated by ContEst16s. Prokka tool for bacterial annotation was used to annotate sequences from both Leptospira isolates. Predicted amino acid sequences from Prokka were searched in EggNOG and David gene ontology database to characterize gene ontology. In addition, Leptospira from mild and severe patients, that passed the criteria e-value < 10e-5 from blastP against virulence factor database, were used to analyze with Venn diagram. From this study, we found 13 and 12 genes that were unique in the isolates from mild and severe patients, respectively. The 12 genes in the severe isolate might be virulence factor genes that affect disease severity. However, these genes should be validated in further study.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
