Daniel K Knewitz, Colin J Anderson, William T Presley, MaryBeth Horodyski, Mark T Scarborough, Margaret R Wallace
{"title":"恶性周围神经鞘肿瘤骨科患者的生存和NF1分析。","authors":"Daniel K Knewitz, Colin J Anderson, William T Presley, MaryBeth Horodyski, Mark T Scarborough, Margaret R Wallace","doi":"10.1155/2021/9386823","DOIUrl":null,"url":null,"abstract":"<p><p>Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor syndrome in which benign plexiform neurofibromas are at risk of transforming into malignant peripheral nerve sheath tumors (MPNSTs), a very rare soft-tissue sarcoma. The prognosis of patients with MPNSTs is poor, with most studies reporting <50% survival at five years. However, studies evaluating MPNSTs are limited and report heterogeneous results. Because no MPNST-specific evidence-based treatment guideline exists, individual institutional experiences are very informative to the field. The main objective of this study was to investigate and report MPNST prognostic clinical and genetic biomarkers from our institution's Orthopedics service experience treating 20 cases from 1992 to 2017. Most patients were treated with resection and adjuvant radiation. Extended follow-up, averaging 11.4 years (ranging 1.1 to 25.1), revealed excellent five-year survival rates: 70% for overall and 60% for metastatic disease. An S100 B immunonegative tumor phenotype was associated with a significantly worse outcome than MPNSTs with positive S100 B stain. In addition, <i>NF1</i> gene mutation analysis was performed on 27 families with NF1 in which at least one affected family member developed MPNSTs. Of the 27 <i>NF1</i> germline mutations, five were large deletions spanning (or nearly spanning) the gene (18.5%), substantially more than such deletions in NF1 in general, consistent with increased risk of MPNSTs in such cases.</p>","PeriodicalId":21431,"journal":{"name":"Sarcoma","volume":"2021 ","pages":"9386823"},"PeriodicalIF":0.0000,"publicationDate":"2021-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8505086/pdf/","citationCount":"2","resultStr":"{\"title\":\"Survival and <i>NF1</i> Analysis in a Cohort of Orthopedics Patients with Malignant Peripheral Nerve Sheath Tumors.\",\"authors\":\"Daniel K Knewitz, Colin J Anderson, William T Presley, MaryBeth Horodyski, Mark T Scarborough, Margaret R Wallace\",\"doi\":\"10.1155/2021/9386823\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor syndrome in which benign plexiform neurofibromas are at risk of transforming into malignant peripheral nerve sheath tumors (MPNSTs), a very rare soft-tissue sarcoma. The prognosis of patients with MPNSTs is poor, with most studies reporting <50% survival at five years. However, studies evaluating MPNSTs are limited and report heterogeneous results. Because no MPNST-specific evidence-based treatment guideline exists, individual institutional experiences are very informative to the field. The main objective of this study was to investigate and report MPNST prognostic clinical and genetic biomarkers from our institution's Orthopedics service experience treating 20 cases from 1992 to 2017. Most patients were treated with resection and adjuvant radiation. Extended follow-up, averaging 11.4 years (ranging 1.1 to 25.1), revealed excellent five-year survival rates: 70% for overall and 60% for metastatic disease. An S100 B immunonegative tumor phenotype was associated with a significantly worse outcome than MPNSTs with positive S100 B stain. In addition, <i>NF1</i> gene mutation analysis was performed on 27 families with NF1 in which at least one affected family member developed MPNSTs. Of the 27 <i>NF1</i> germline mutations, five were large deletions spanning (or nearly spanning) the gene (18.5%), substantially more than such deletions in NF1 in general, consistent with increased risk of MPNSTs in such cases.</p>\",\"PeriodicalId\":21431,\"journal\":{\"name\":\"Sarcoma\",\"volume\":\"2021 \",\"pages\":\"9386823\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-10-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8505086/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Sarcoma\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2021/9386823\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Sarcoma","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2021/9386823","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Medicine","Score":null,"Total":0}
Survival and NF1 Analysis in a Cohort of Orthopedics Patients with Malignant Peripheral Nerve Sheath Tumors.
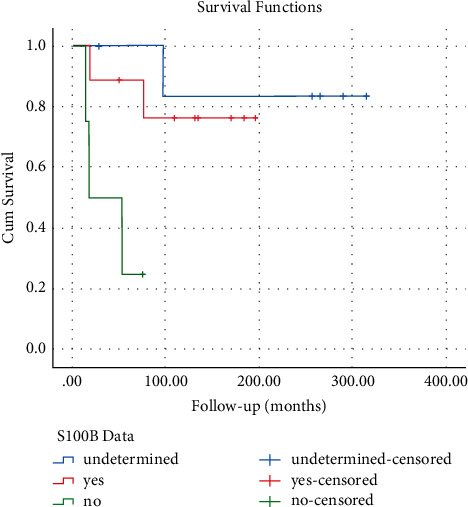
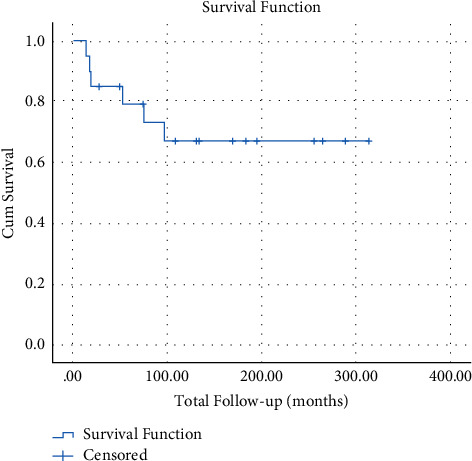
Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor syndrome in which benign plexiform neurofibromas are at risk of transforming into malignant peripheral nerve sheath tumors (MPNSTs), a very rare soft-tissue sarcoma. The prognosis of patients with MPNSTs is poor, with most studies reporting <50% survival at five years. However, studies evaluating MPNSTs are limited and report heterogeneous results. Because no MPNST-specific evidence-based treatment guideline exists, individual institutional experiences are very informative to the field. The main objective of this study was to investigate and report MPNST prognostic clinical and genetic biomarkers from our institution's Orthopedics service experience treating 20 cases from 1992 to 2017. Most patients were treated with resection and adjuvant radiation. Extended follow-up, averaging 11.4 years (ranging 1.1 to 25.1), revealed excellent five-year survival rates: 70% for overall and 60% for metastatic disease. An S100 B immunonegative tumor phenotype was associated with a significantly worse outcome than MPNSTs with positive S100 B stain. In addition, NF1 gene mutation analysis was performed on 27 families with NF1 in which at least one affected family member developed MPNSTs. Of the 27 NF1 germline mutations, five were large deletions spanning (or nearly spanning) the gene (18.5%), substantially more than such deletions in NF1 in general, consistent with increased risk of MPNSTs in such cases.
SarcomaMedicine-Radiology, Nuclear Medicine and Imaging
CiteScore
5.00
自引率
0.00%
发文量
15
审稿时长
14 weeks
期刊介绍:
Sarcoma is dedicated to publishing papers covering all aspects of connective tissue oncology research. It brings together work from scientists and clinicians carrying out a broad range of research in this field, including the basic sciences, molecular biology and pathology and the clinical sciences of epidemiology, surgery, radiotherapy and chemotherapy. High-quality papers concerning the entire range of bone and soft tissue sarcomas in both adults and children, including Kaposi"s sarcoma, are published as well as preclinical and animal studies. This journal provides a central forum for the description of advances in diagnosis, assessment and treatment of this rarely seen, but often mismanaged, group of patients.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
